Paedriatic airway atlas: all Tissues
Quality control: Droplet processing with emptyDroplets
Gunjan Dixit and Jovana Maksimovic
August 15, 2023
Last updated: 2023-08-15
Checks: 7 0
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version baf913b. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/RDS/
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Untracked files:
Untracked: figure/
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/01_QC_emptyDrops.Rmd) and
HTML (docs/01_QC_emptyDrops.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 5085c7b | Gunjan Dixit | 2023-08-11 | Build site. |
| html | 1a72faa | Gunjan Dixit | 2023-08-11 | Build site. |
| Rmd | 0bdb3ba | Gunjan Dixit | 2023-08-11 | wflow_publish("analysis/*") |
INTRODUCTION
This RMarkdown reads CellRanger output RDS file for each batch and identifies empty-droplets in the data followed by the their removal. Empty droplets can contain ambient (i.e., extracellular) RNA that can be captured and sequenced, resulting in non-zero counts for libraries that do not contain any cell.
Batch 1- Nasal brushings (n=16)
Batch
2_1- Tonsils (n=16)
Batch 2_2- Nasal
brushings (n=9) (repeats)
Batch 3- Adenoids (n=16)
Batch 4- Bronchial brushings (n=16)
Batch 5- Nasal brushings (n=16)
Load libraries
suppressPackageStartupMessages({
library(BiocStyle)
library(tidyverse)
library(here)
library(dplyr)
library(glue)
library(DropletUtils)
library(kableExtra)
})List batch files
files <- list.files(here("data",
"RDS"),
pattern = "CellRanger",
full.names = TRUE)
tissue_types <- c("Nasal_brushings", "Tonsils", "Nasal_brushings_repeat", "Adenoids", "Bronchial_brushings", "Nasal_brushings_2")
batches <- sub("\\.CellRanger\\.SCE\\.rds$", "", basename(files))
batches[1] "G000231_Neeland_batch1" "G000231_Neeland_batch2_1"
[3] "G000231_Neeland_batch2_2" "G000231_Neeland_batch3"
[5] "G000231_Neeland_batch4" "G000231_Neeland_batch5" Create Output dirs
# Create Output dirs
sapply(1:length(batches), function(i){
dir.create(here("output", batches[i], "RDS", "SCEs"), recursive = TRUE)
dir.create(here("output", batches[i], "emptyDrops"))
})[1] FALSE FALSE FALSE FALSE FALSE FALSEIdentify empty droplets
Run emptydrops on each batch and sample individually to identify
empty droplets. We use the emptyDrops() function from the
DropletUtils
package to test whether the expression profile for each cell barcode is
significantly different from the ambient RNA pool [@lun2018distinguishing]. A significant
deviation indicates that the barcode corresponds to a cell-containing
droplet. Cells are called at a false discovery rate (FDR) of 0.1%.
# Function to run emptyDrops and save output for a batch
process_batch_emptyDrops <- function(batch_name) {
file <- files[grepl(batch_name, files)]
sce <- readRDS(file)
for (s in unique(sce$Sample)) {
sce_sample <- sce[, sce$Sample == s]
output_dir <- here("output", batch_name, "emptyDrops")
out <- file.path(output_dir, paste0(s, ".emptyDrops.rds"))
if (file.exists(out)) {
next # Skip this sample and move to the next one
}
e.out <- emptyDrops(counts(sce_sample))
saveRDS(e.out, file = out)
}
}
# Loop through each batch
for (batch_name in batches) {
process_batch_emptyDrops(batch_name)
}Read RDS files
Read emptyDrops (droplets processed) RDS files for each batch and sample.Remove empty droplets by subsetting SingleCellExperiment (SCE) object to retain only the detected cells. which() removes the NAs prior to the subsetting. Remove genes that are not expressed in any cells and save the SCE object.
all_non_empty_droplets <- list()
# Create an empty data frame to store the master table
master_table <- data.frame(Batch = character(), Sample = character(), Non_Empty_Droplets = numeric(), stringsAsFactors = FALSE)
for (batch_name in unique(batches)) {
file <- files[grepl(batch_name, files)]
sce <- readRDS(file)
batch_files <- list.files(
path = here("output", batch_name, "emptyDrops"),
pattern = "\\.emptyDrops\\.rds$",
full.names = TRUE
)
# Extract sample names from the file names using the provided pattern
sample_names <- gsub(".emptyDrops.rds", "", basename(batch_files))
empties_list <- list()
for (sn in sample_names) {
empties <- readRDS(
here("output", batch_name, "emptyDrops", paste0(sn, ".emptyDrops.rds"))
)
empties$sample <- sn
empties_list[[sn]] <- empties
}
# Combine the data for all samples in this batch into a single data frame
combined_data <- do.call(rbind, empties_list)
# Remove empty droplets based on FDR threshold
sce <- sce[, which(combined_data$FDR <= 0.001)]
# Remove genes not expressed in any cells
sce <- sce[rowSums(counts(sce)) > 0,]
# Save the output SingleCellExperiment object
out <- here("output", batch_name, "RDS", "SCEs", paste0(batch_name, ".emptyDrops.SCE.rds"))
if (!file.exists(out)) saveRDS(sce, file = out)
# Calculate the number of non-empty droplets for each sample in this batch
non_empty_droplets <- tapply(
combined_data$FDR,
combined_data$sample,
function(x) sum(x <= 0.001, na.rm = TRUE)
)
# Store the non-empty counts for this batch in the list
all_non_empty_droplets[[batch_name]] <- non_empty_droplets
# Create a data frame for this batch with Batch, Sample, and Non_Empty_Count columns
batch_table <- data.frame(Batch = batch_name, Sample = names(non_empty_droplets), Non_Empty_Droplets = unlist(non_empty_droplets), stringsAsFactors = FALSE)
# Append the batch_table to the master_table
master_table <- rbind(master_table, batch_table)
}Examine number of non-empty droplets
Table with number of non-empty droplets for each sample in each batch.
for (i in seq_along(batches)){
batch_name <- batches[i]
tissue_type <- tissue_types[i]
cat(paste0("## ", batch_name, " - ", tissue_type, "\n"))
print(kable(master_table[master_table$Batch == batch_name, ],
caption = paste("Table of non-empty droplets for", tissue_type),
format = "html",
row.names = FALSE) %>% kable_styling())
cat("\n\n")
}G000231_Neeland_batch1 - Nasal_brushings
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch1 | eAIR004 | 4161 |
| G000231_Neeland_batch1 | eAIR005 | 865 |
| G000231_Neeland_batch1 | eAIR006 | 3358 |
| G000231_Neeland_batch1 | eAIR007 | 1818 |
| G000231_Neeland_batch1 | eAIR011 | 3764 |
| G000231_Neeland_batch1 | eAIR013 | 3880 |
| G000231_Neeland_batch1 | eAIR016 | 3496 |
| G000231_Neeland_batch1 | eAIR017 | 1641 |
| G000231_Neeland_batch1 | eAIR019 | 3834 |
| G000231_Neeland_batch1 | eAIR020 | 2723 |
| G000231_Neeland_batch1 | eAIR024 | 4972 |
| G000231_Neeland_batch1 | eAIR025 | 4224 |
| G000231_Neeland_batch1 | eAIR027 | 5528 |
| G000231_Neeland_batch1 | eAIR028 | 6603 |
| G000231_Neeland_batch1 | eAIR031 | 2664 |
| G000231_Neeland_batch1 | eAIR032 | 3161 |
G000231_Neeland_batch2_1 - Tonsils
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch2_1 | eAIR002 | 10834 |
| G000231_Neeland_batch2_1 | eAIR004 | 10471 |
| G000231_Neeland_batch2_1 | eAIR006 | 11549 |
| G000231_Neeland_batch2_1 | eAIR007 | 11645 |
| G000231_Neeland_batch2_1 | eAIR008 | 11428 |
| G000231_Neeland_batch2_1 | eAIR010 | 12929 |
| G000231_Neeland_batch2_1 | eAIR012 | 8883 |
| G000231_Neeland_batch2_1 | eAIR013 | 13132 |
| G000231_Neeland_batch2_1 | eAIR014 | 9746 |
| G000231_Neeland_batch2_1 | eAIR016 | 13110 |
| G000231_Neeland_batch2_1 | eAIR017 | 12831 |
| G000231_Neeland_batch2_1 | eAIR018 | 11104 |
| G000231_Neeland_batch2_1 | eAIR019 | 10697 |
| G000231_Neeland_batch2_1 | eAIR020 | 10942 |
| G000231_Neeland_batch2_1 | eAIR021 | 12008 |
| G000231_Neeland_batch2_1 | eAIR022 | 9904 |
G000231_Neeland_batch2_2 - Nasal_brushings_repeat
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch2_2 | eAIR004 | 16757 |
| G000231_Neeland_batch2_2 | eAIR016 | 19549 |
| G000231_Neeland_batch2_2 | eAIR019 | 12147 |
| G000231_Neeland_batch2_2 | eAIR020 | 13272 |
| G000231_Neeland_batch2_2 | eAIR024 | 22580 |
| G000231_Neeland_batch2_2 | eAIR025 | 22375 |
| G000231_Neeland_batch2_2 | eAIR028 | 27573 |
| G000231_Neeland_batch2_2 | eAIR031 | 2575 |
| G000231_Neeland_batch2_2 | eAIR032 | 7829 |
G000231_Neeland_batch3 - Adenoids
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch3 | eAIR001 | 5954 |
| G000231_Neeland_batch3 | eAIR003 | 5835 |
| G000231_Neeland_batch3 | eAIR004 | 6008 |
| G000231_Neeland_batch3 | eAIR006 | 6424 |
| G000231_Neeland_batch3 | eAIR007 | 8121 |
| G000231_Neeland_batch3 | eAIR008 | 6046 |
| G000231_Neeland_batch3 | eAIR010 | 7768 |
| G000231_Neeland_batch3 | eAIR012 | 6225 |
| G000231_Neeland_batch3 | eAIR013 | 6977 |
| G000231_Neeland_batch3 | eAIR014 | 4920 |
| G000231_Neeland_batch3 | eAIR015 | 7867 |
| G000231_Neeland_batch3 | eAIR016 | 7706 |
| G000231_Neeland_batch3 | eAIR019 | 11545 |
| G000231_Neeland_batch3 | eAIR020 | 8249 |
| G000231_Neeland_batch3 | eAIR021 | 6014 |
| G000231_Neeland_batch3 | eAIR023 | 6132 |
G000231_Neeland_batch4 - Bronchial_brushings
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch4 | eAIR009 | 4187 |
| G000231_Neeland_batch4 | eAIR010 | 2245 |
| G000231_Neeland_batch4 | eAIR012 | 1910 |
| G000231_Neeland_batch4 | eAIR013 | 4551 |
| G000231_Neeland_batch4 | eAIR014 | 4046 |
| G000231_Neeland_batch4 | eAIR016 | 6652 |
| G000231_Neeland_batch4 | eAIR018 | 1655 |
| G000231_Neeland_batch4 | eAIR020 | 4153 |
| G000231_Neeland_batch4 | eAIR021 | 2923 |
| G000231_Neeland_batch4 | eAIR022 | 4866 |
| G000231_Neeland_batch4 | eAIR024 | 3930 |
| G000231_Neeland_batch4 | eAIR025 | 1863 |
| G000231_Neeland_batch4 | eAIR026 | 6784 |
| G000231_Neeland_batch4 | eAIR027 | 4827 |
| G000231_Neeland_batch4 | eAIR028 | 2298 |
| G000231_Neeland_batch4 | eAIR031 | 4530 |
G000231_Neeland_batch5 - Nasal_brushings_2
| Batch | Sample | Non_Empty_Droplets |
|---|---|---|
| G000231_Neeland_batch5 | eAIR003 | 3448 |
| G000231_Neeland_batch5 | eAIR008 | 2265 |
| G000231_Neeland_batch5 | eAIR009 | 8901 |
| G000231_Neeland_batch5 | eAIR010 | 4037 |
| G000231_Neeland_batch5 | eAIR014 | 7731 |
| G000231_Neeland_batch5 | eAIR018 | 7588 |
| G000231_Neeland_batch5 | eAIR021 | 6572 |
| G000231_Neeland_batch5 | eAIR022 | 8554 |
| G000231_Neeland_batch5 | eAIR023 | 4709 |
| G000231_Neeland_batch5 | eAIR026 | 8557 |
| G000231_Neeland_batch5 | eAIR030 | 3532 |
| G000231_Neeland_batch5 | eAIR033 | 7760 |
| G000231_Neeland_batch5 | eAIR037 | 6715 |
| G000231_Neeland_batch5 | eAIR038 | 5735 |
| G000231_Neeland_batch5 | eAIR042 | 6486 |
| G000231_Neeland_batch5 | eAIR047 | 5920 |
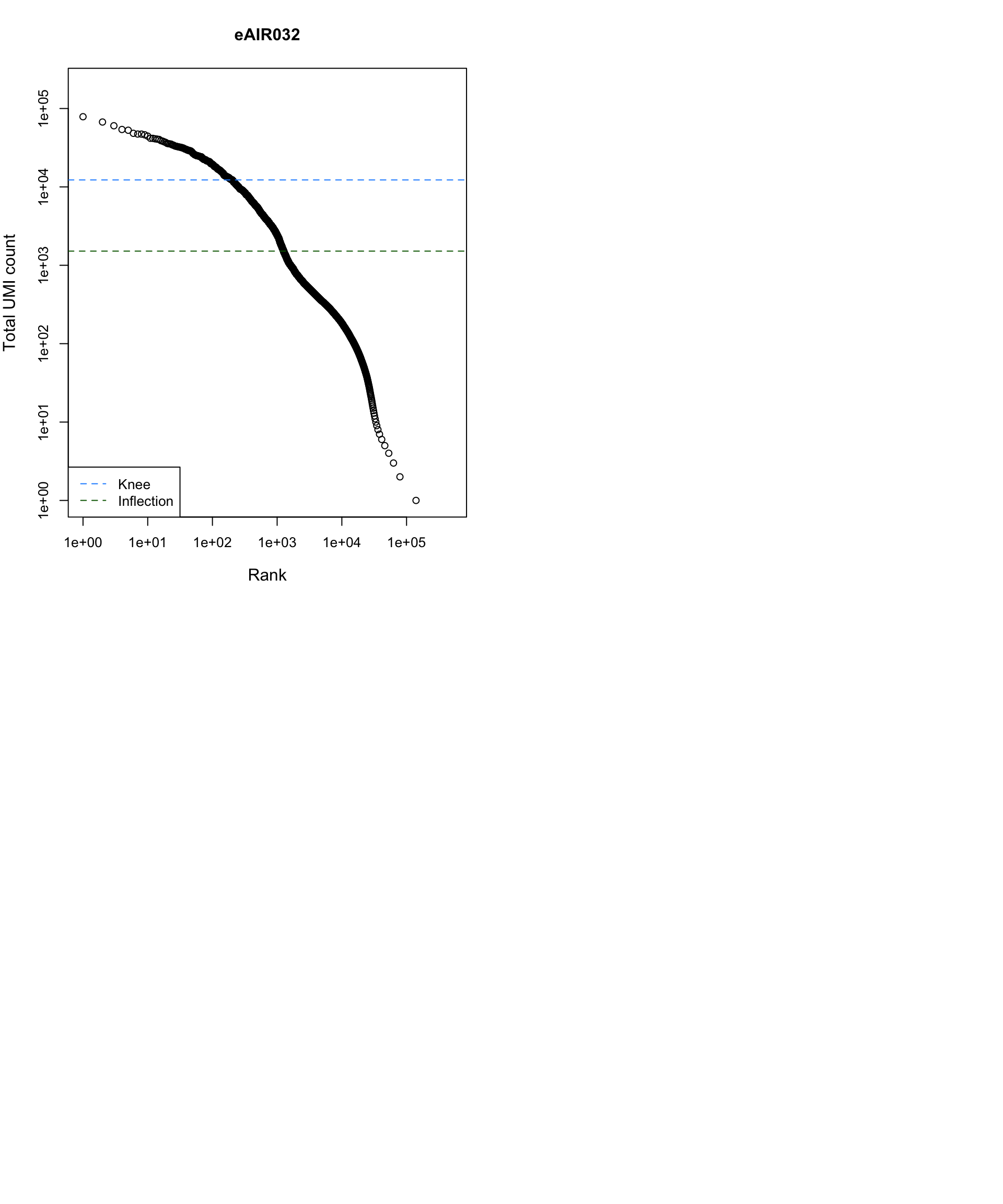
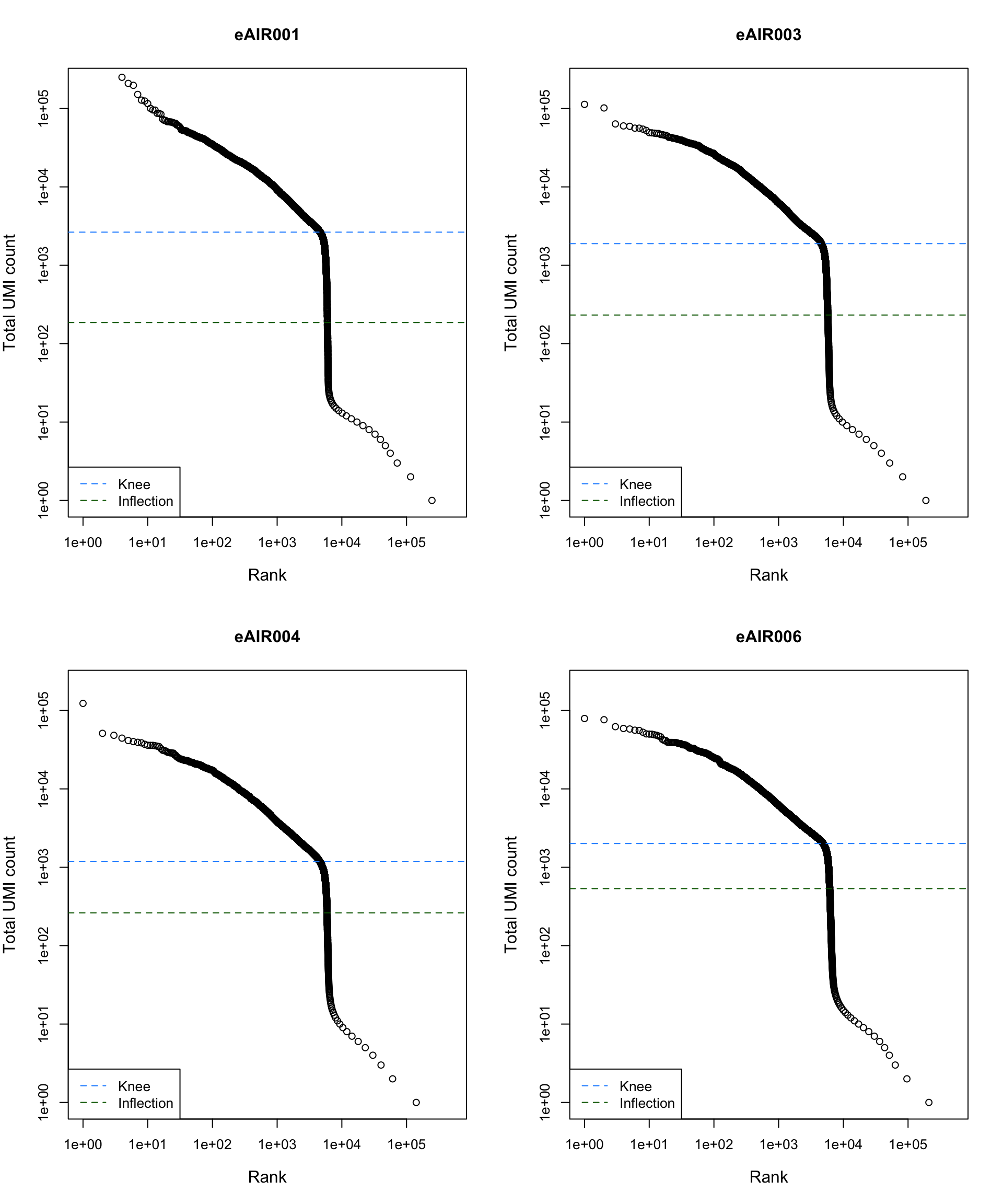
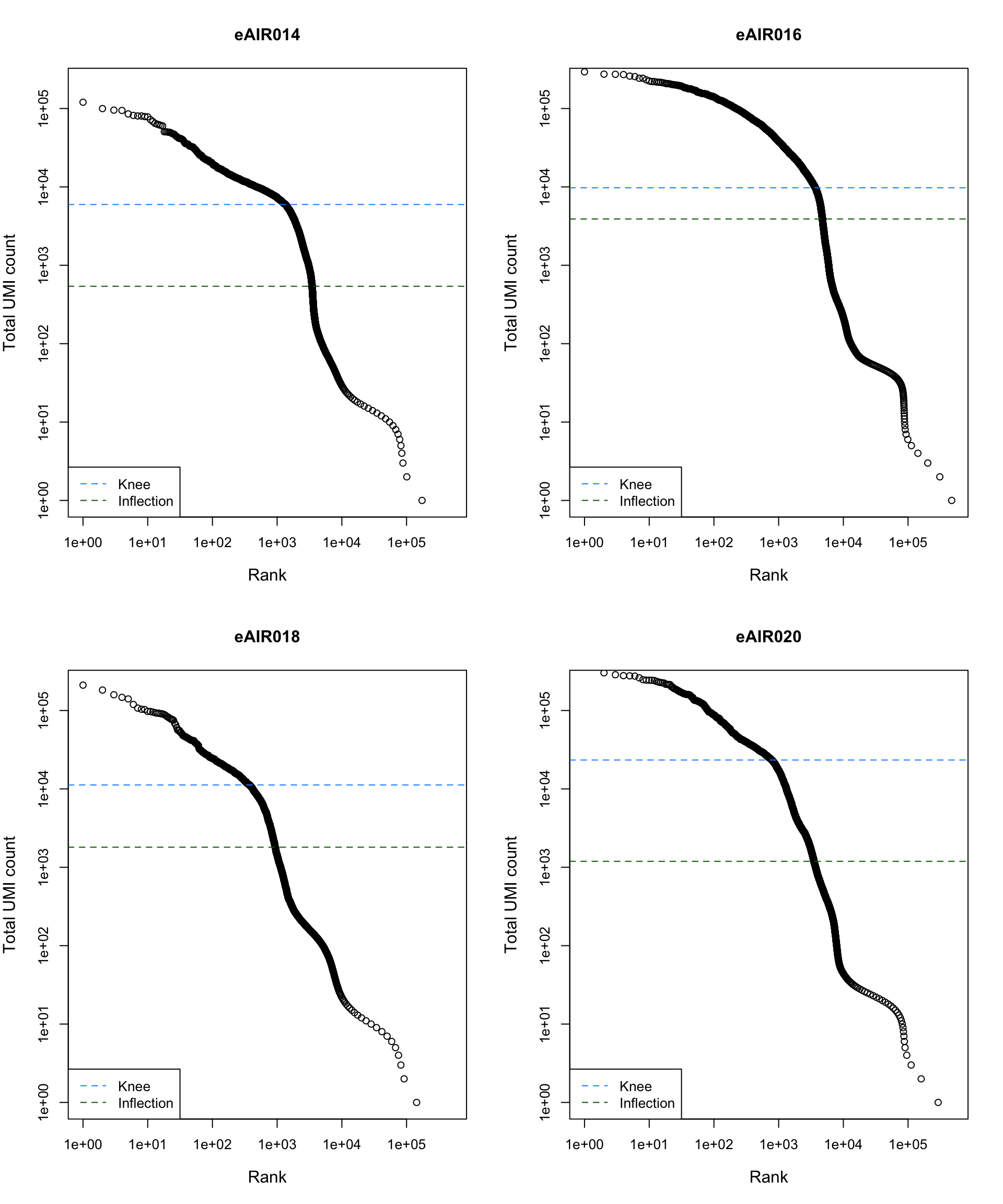
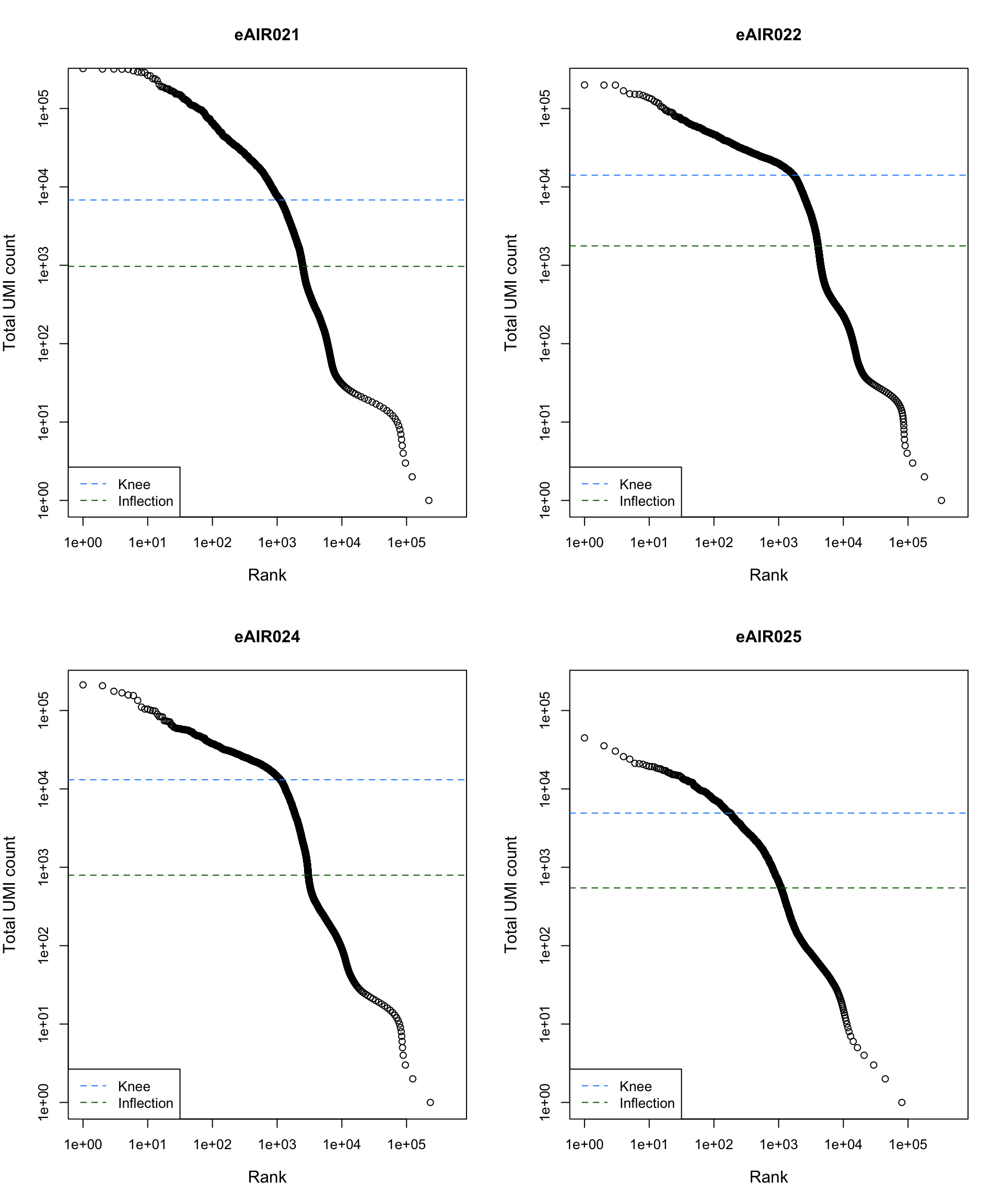
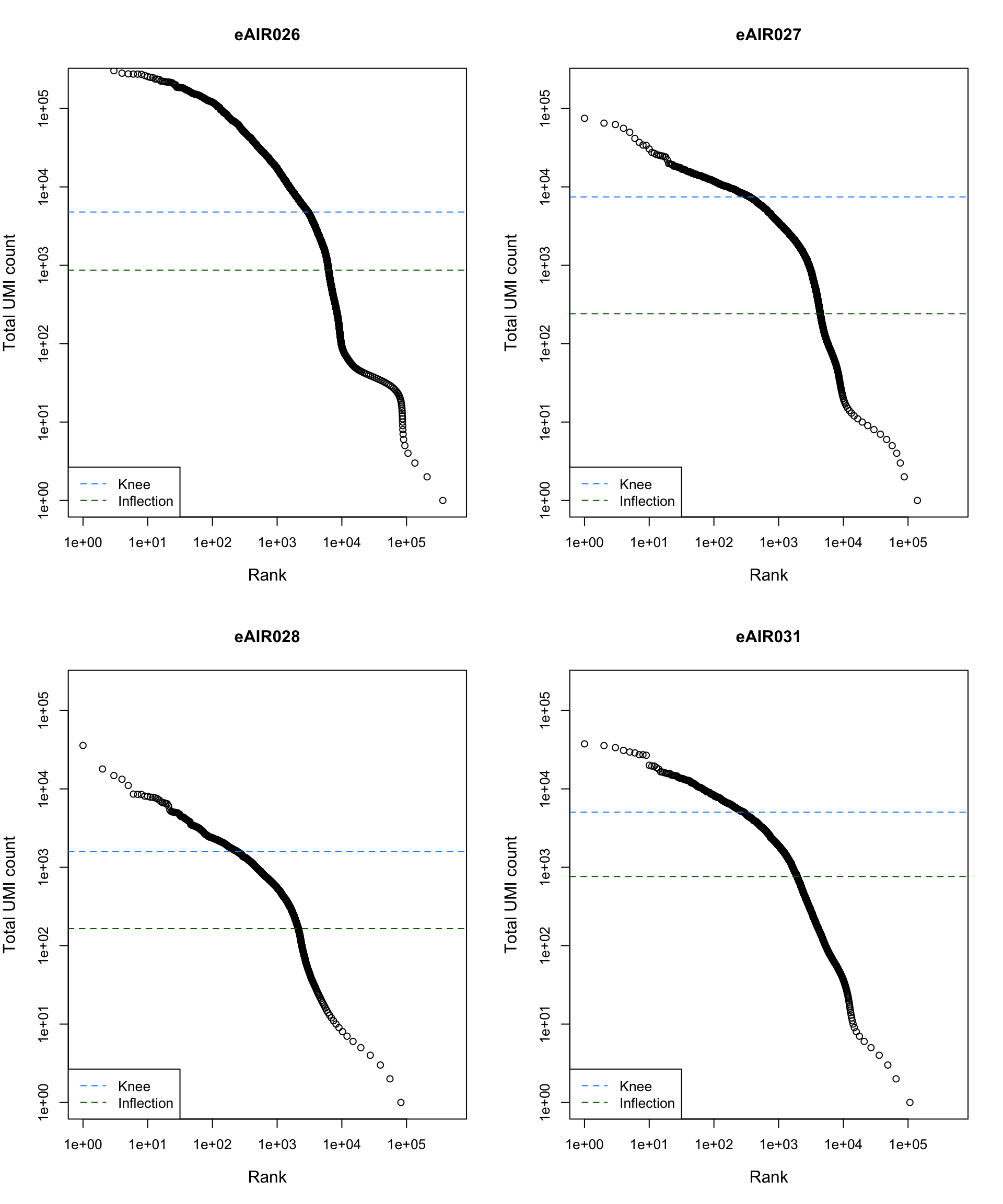
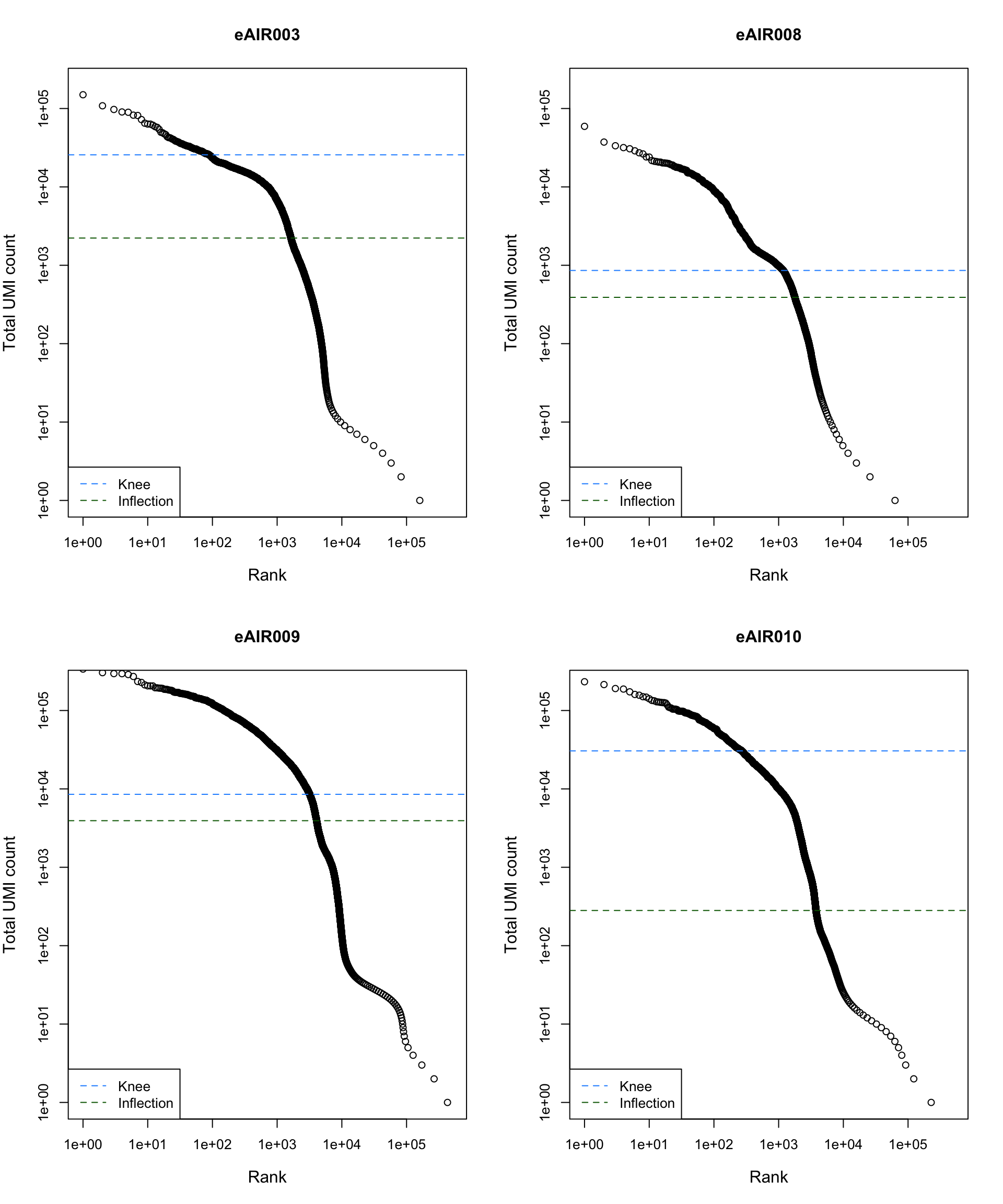
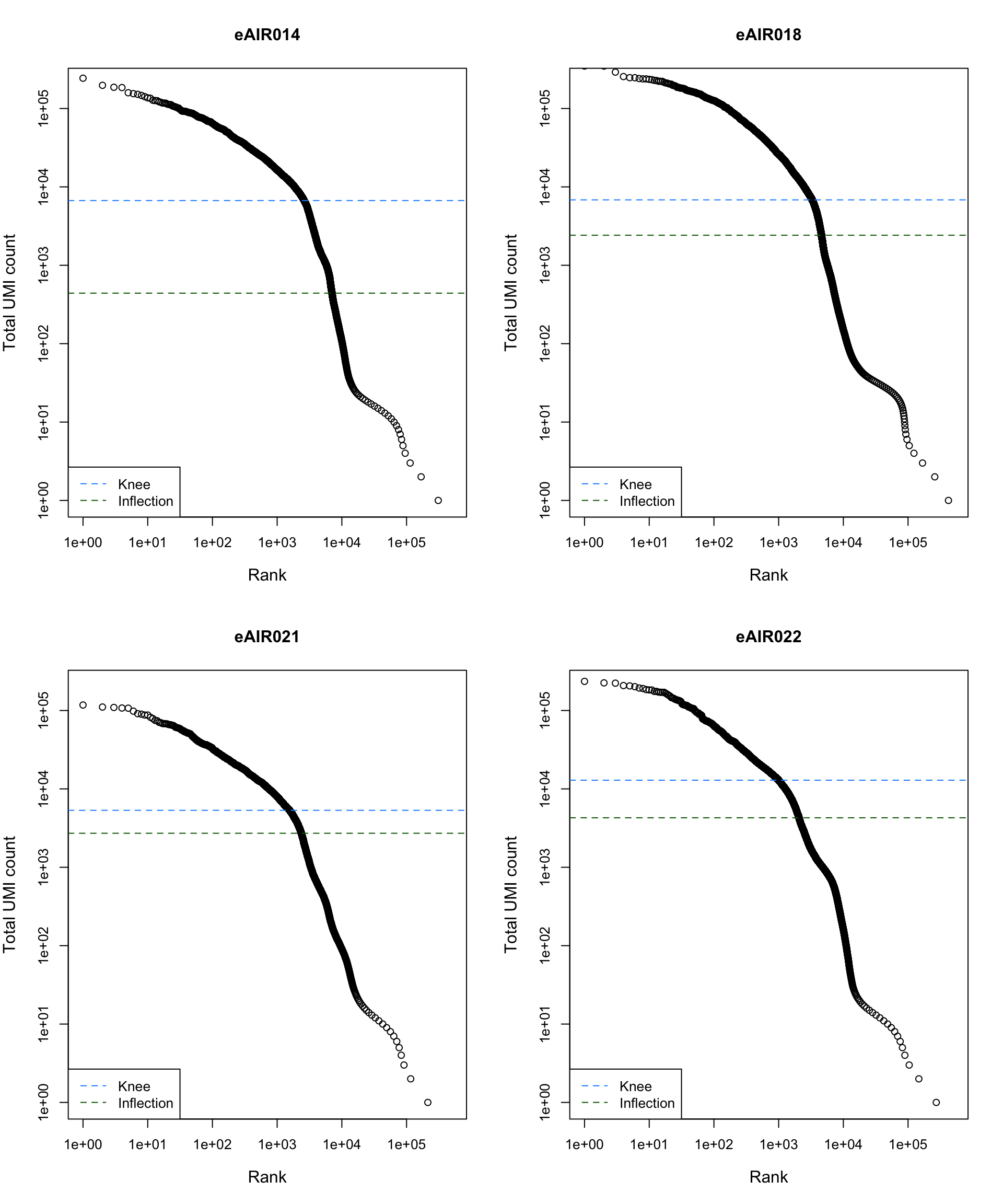
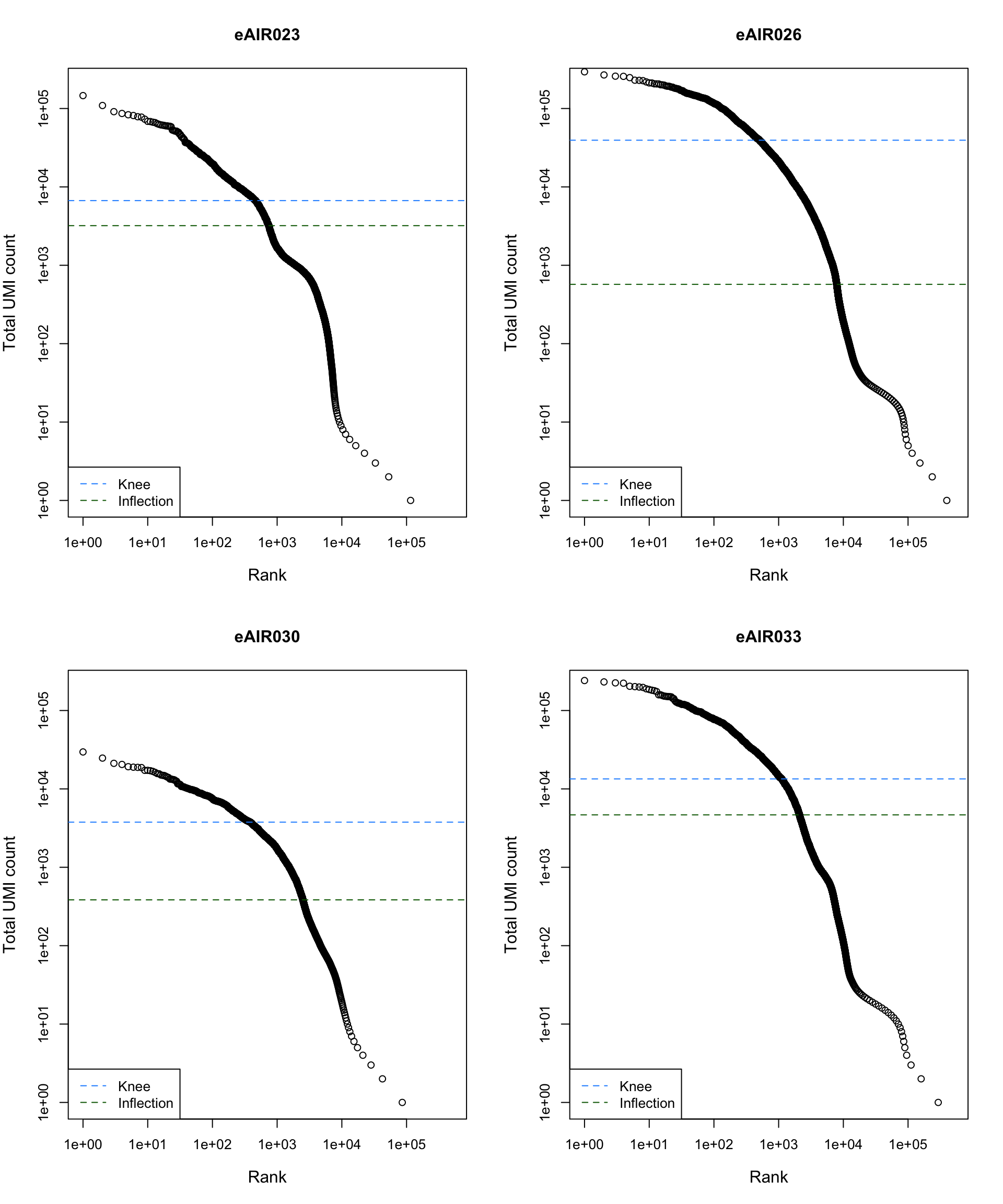
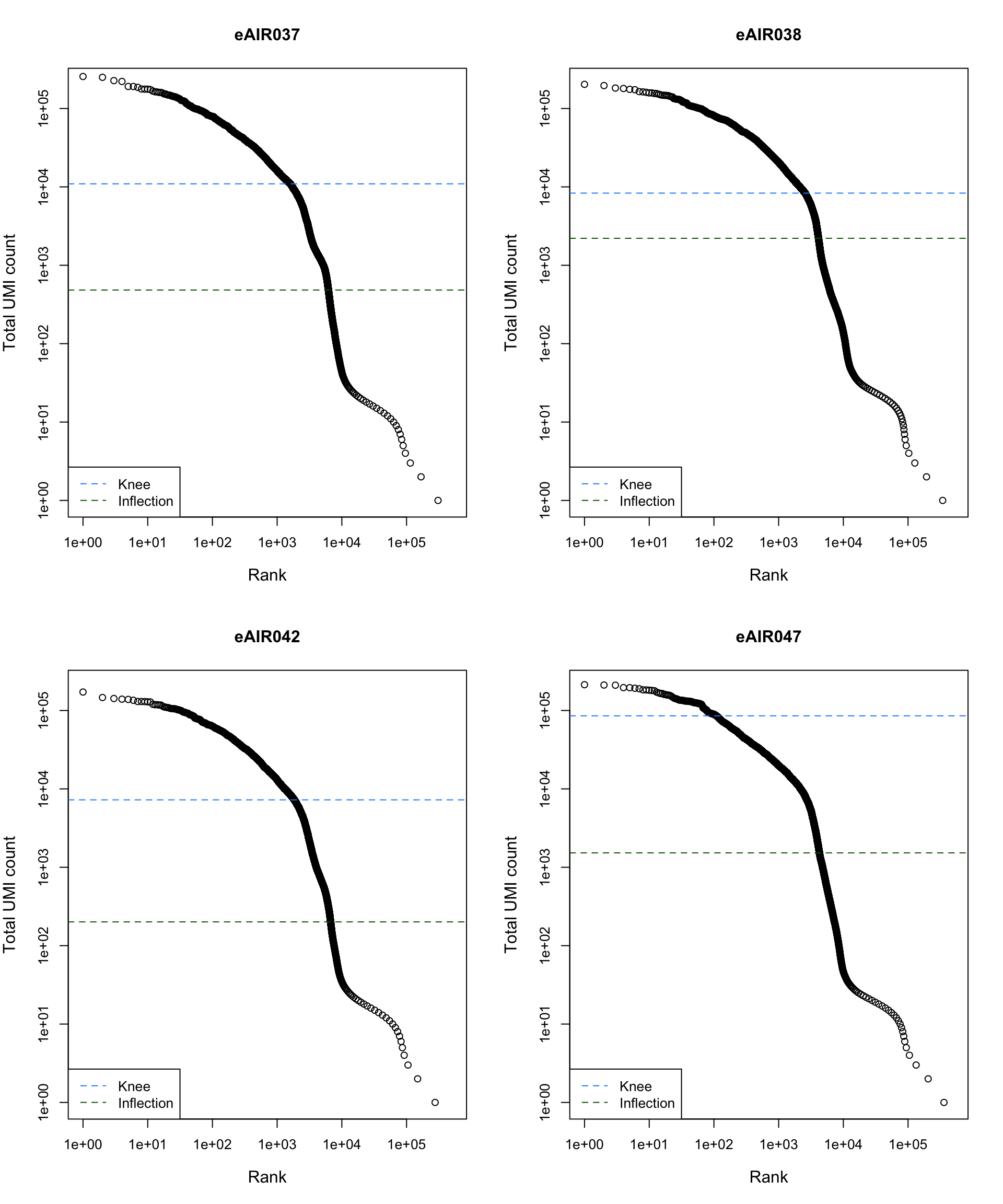
Barcode Rank Plot
Barcode rank plot shows the (log-)total UMI count for each barcode on the y-axis and the (log-)rank on the x-axis. This is effectively a transposed empirical cumulative density plot with log-transformed axes. It examines the distribution of total counts across barcodes, focusing on those with the largest counts.
The knee and inflection points on the curve mark the transition
between two components of the total count distribution. This is assumed
to represent the difference between empty droplets with little RNA and
cell-containing droplets with much more RNA.
Plot barcode rank plots for each sample
# Function to process batch data and plot barcode rank for each sample
process_batch_and_plot <- function(batch_name) {
file <- files[grepl(batch_name, files)]
sce <- readRDS(file)
par(mfrow = c(2, 2))
for (s in unique(sce$Sample)) {
sce_sample <- sce[, sce$Sample == s]
plot_barcode_rank(sce_sample, s)
}
}
# Function to plot barcode rank for each sample
plot_barcode_rank <- function(sample_data, sample_name) {
bcrank <- barcodeRanks(counts(sample_data))
# Only showing unique points for plotting speed.
uniq <- !duplicated(bcrank$rank)
plot(
x = bcrank$rank[uniq],
y = bcrank$total[uniq],
log = "xy",
xlab = "Rank",
ylab = "Total UMI count",
main = sample_name,
cex.lab = 1.2,
xlim = c(1, 500000),
ylim = c(1, 200000)
)
abline(h = metadata(bcrank)$inflection, col = "darkgreen", lty = 2)
abline(h = metadata(bcrank)$knee, col = "dodgerblue", lty = 2)
legend("bottomleft", legend = c("Knee", "Inflection"),
col = c("dodgerblue", "darkgreen"), lty = 2, cex = 1)
}
# Loop through each batch and process the samples
for (i in seq_along(batches)){
batch_name <- batches[i]
tissue_type <- tissue_types[i]
cat('### ', batch_name, " - ", tissue_type, '\n')
print(batch_name)
process_batch_and_plot(batch_name)
cat('\n\n')
}G000231_Neeland_batch1 - Nasal_brushings
[1] “G000231_Neeland_batch1”
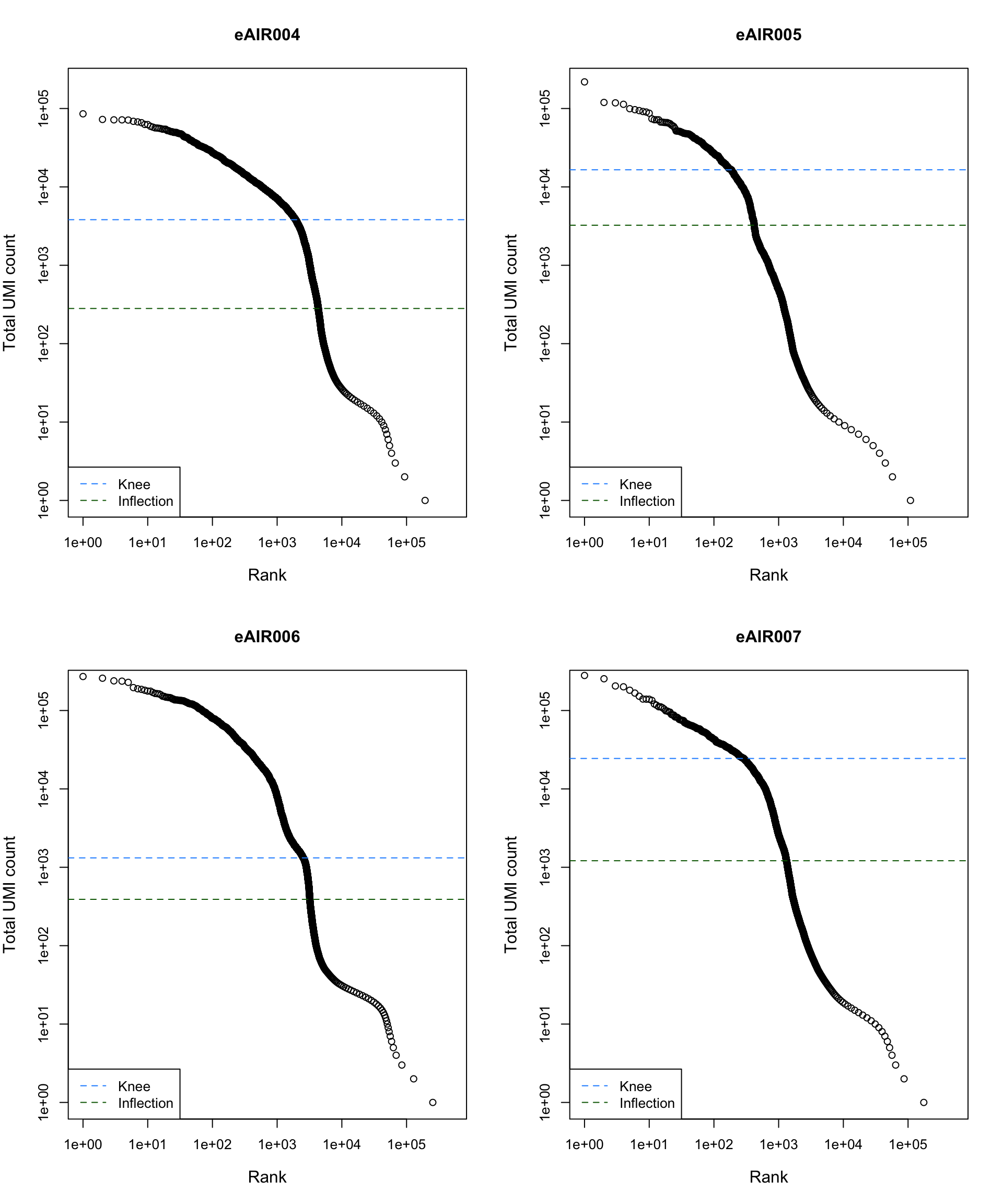
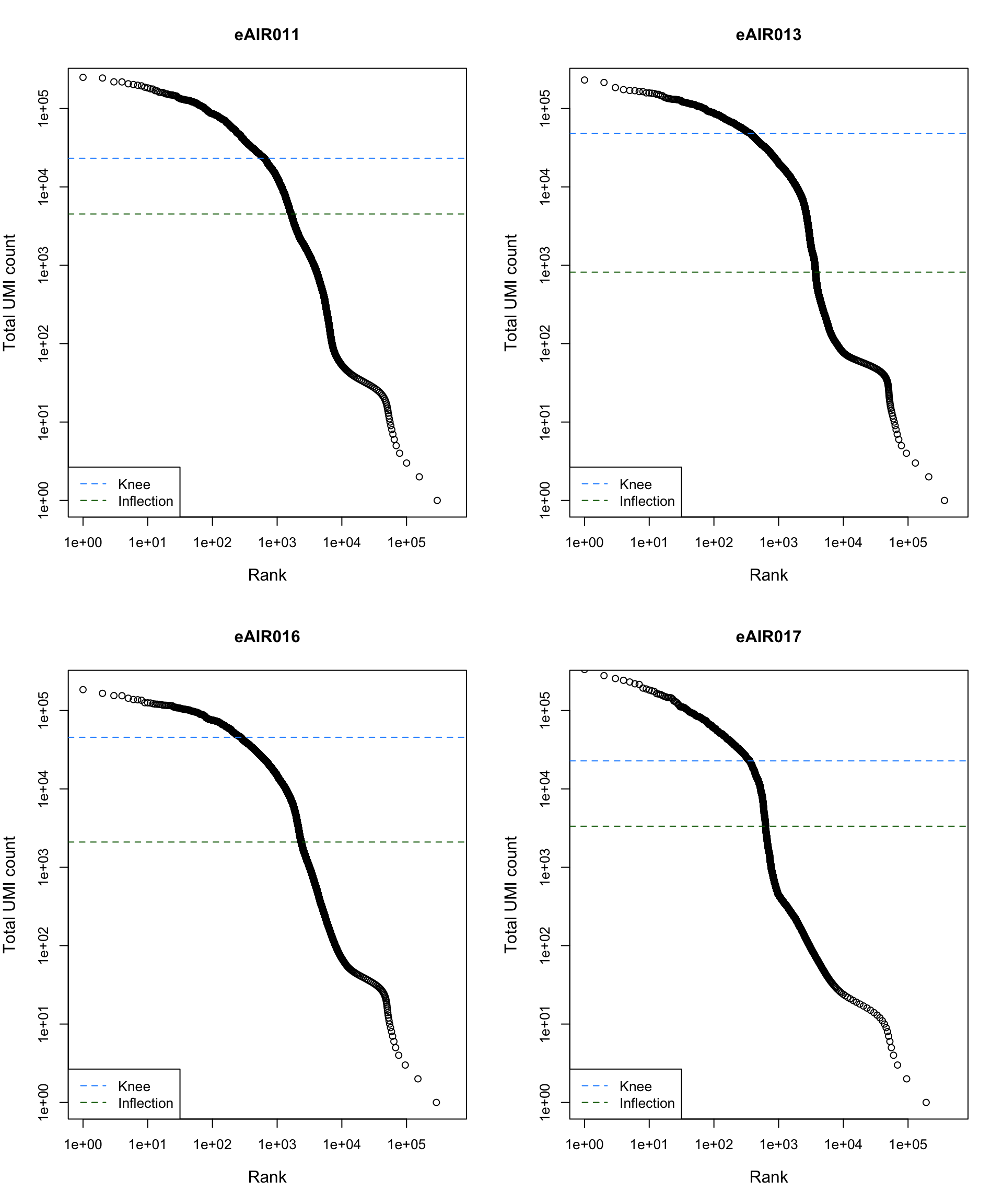
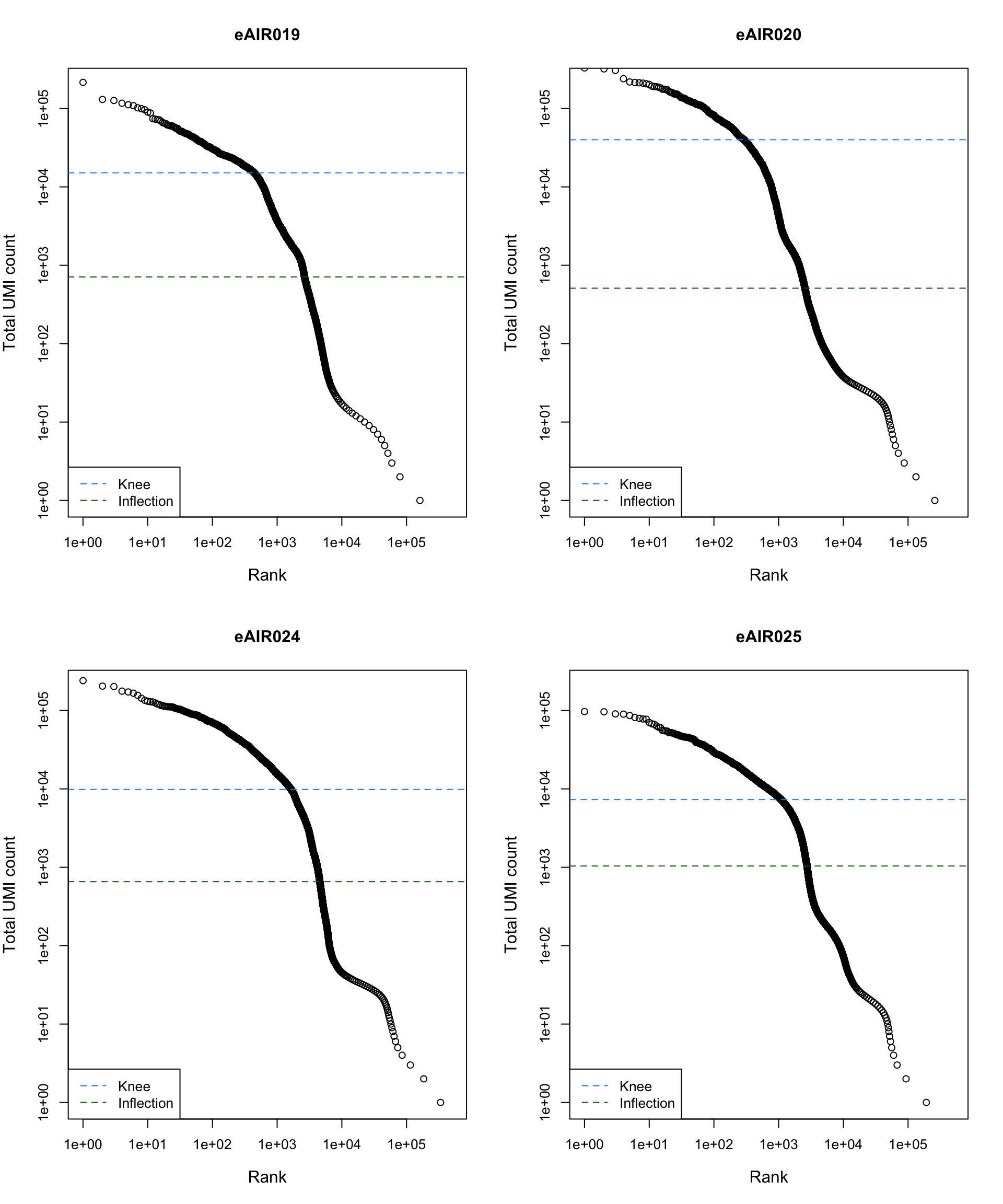
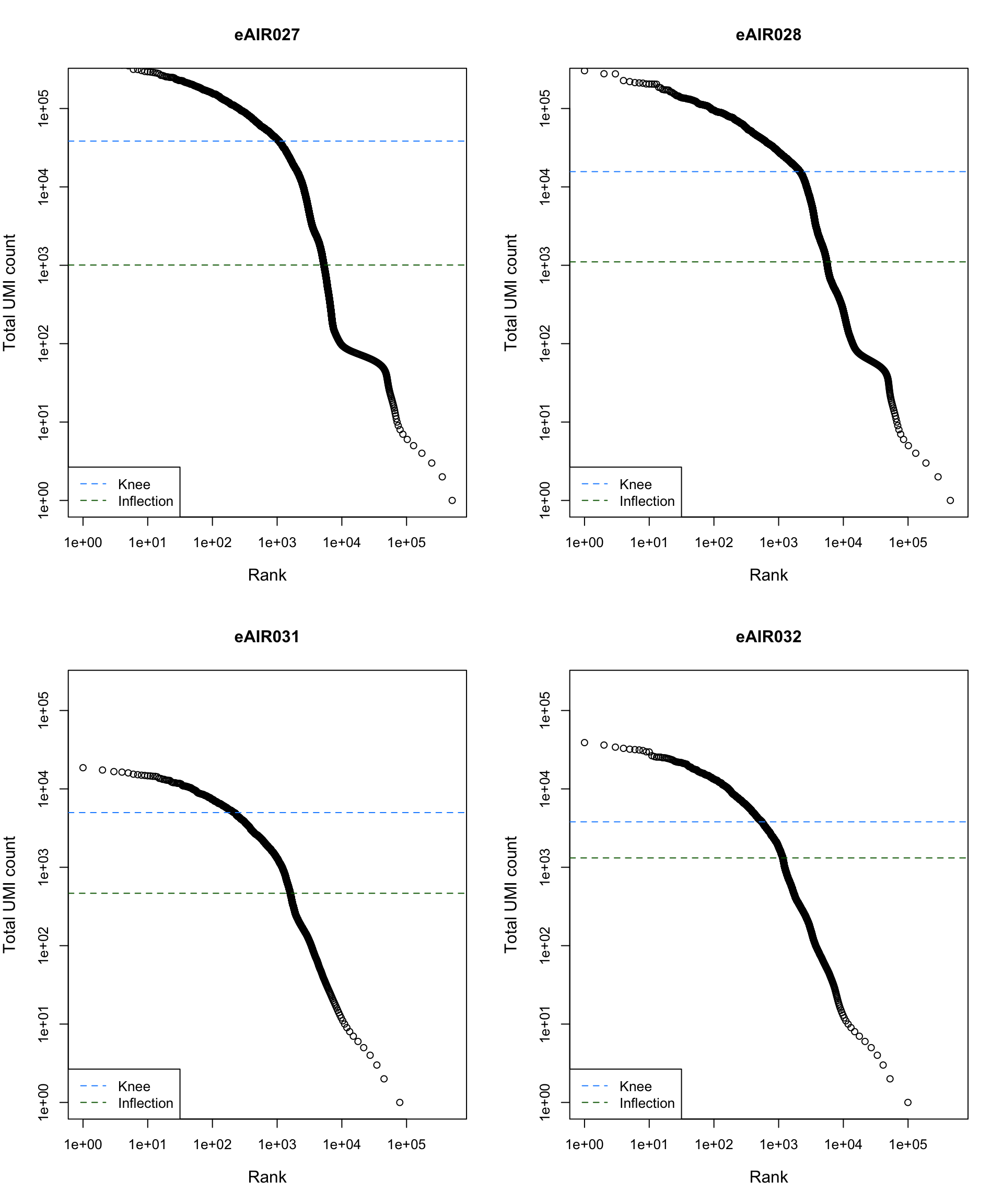
G000231_Neeland_batch2_1 - Tonsils
[1] “G000231_Neeland_batch2_1”
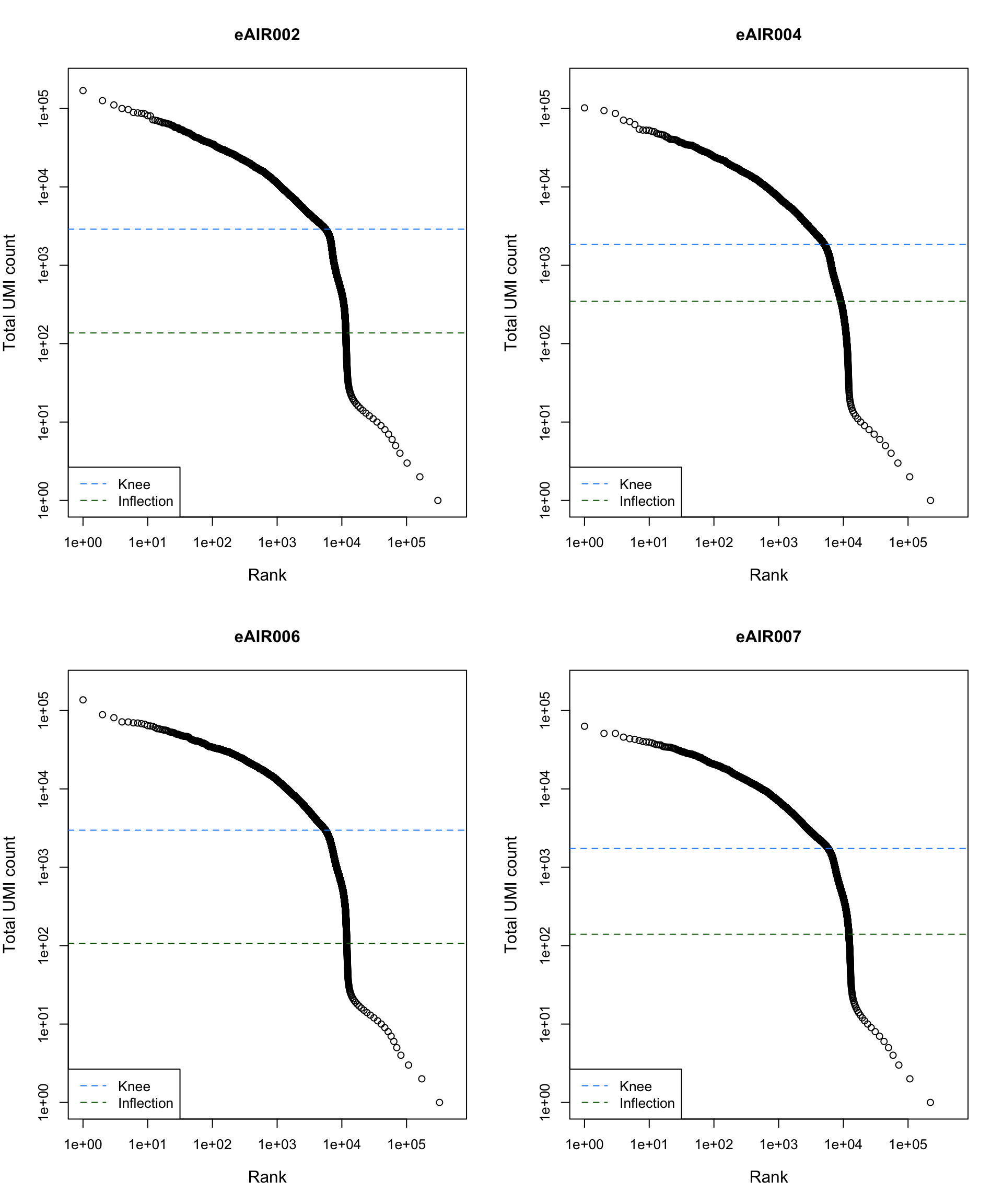
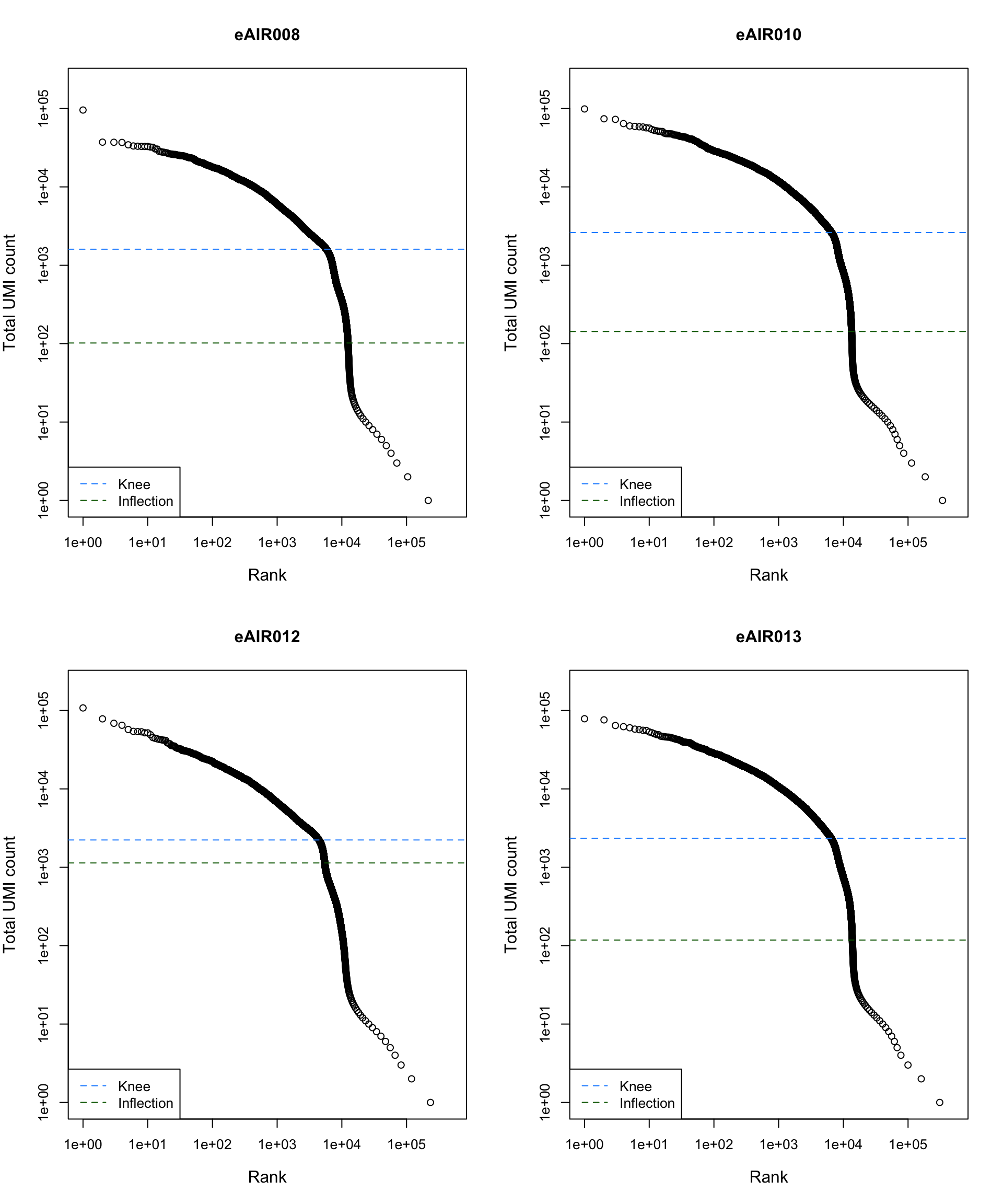
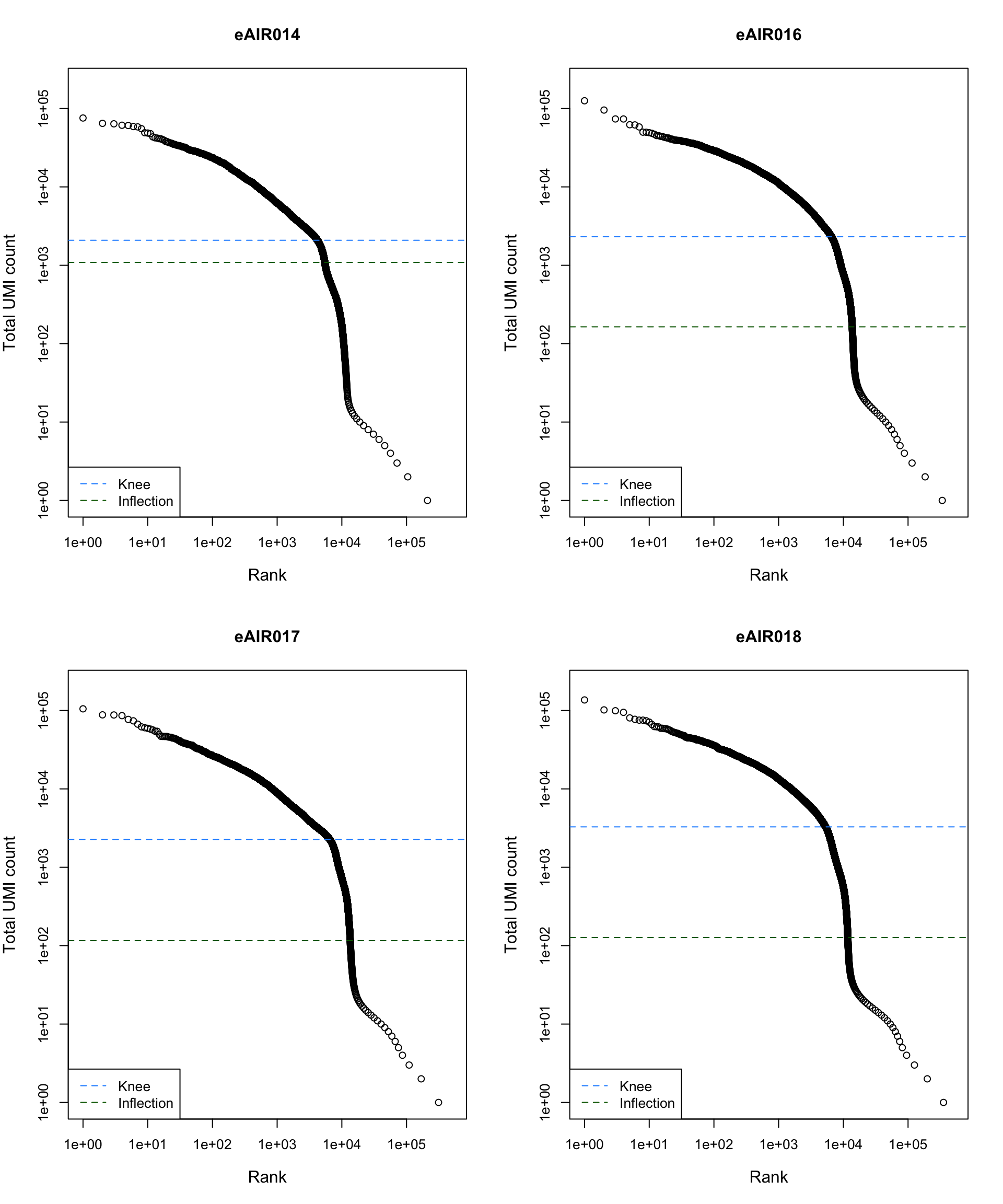
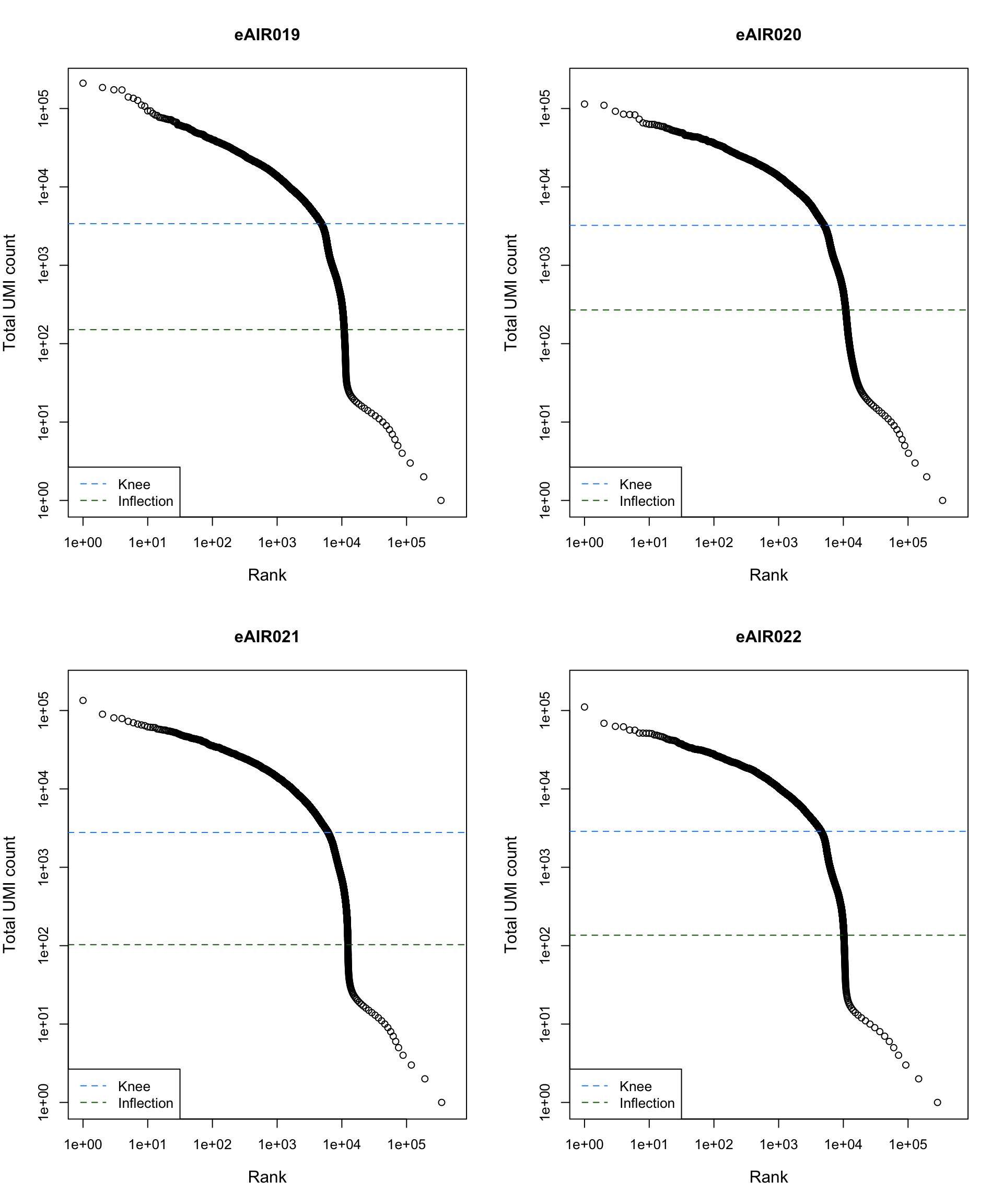
G000231_Neeland_batch2_2 - Nasal_brushings_repeat
[1] “G000231_Neeland_batch2_2”
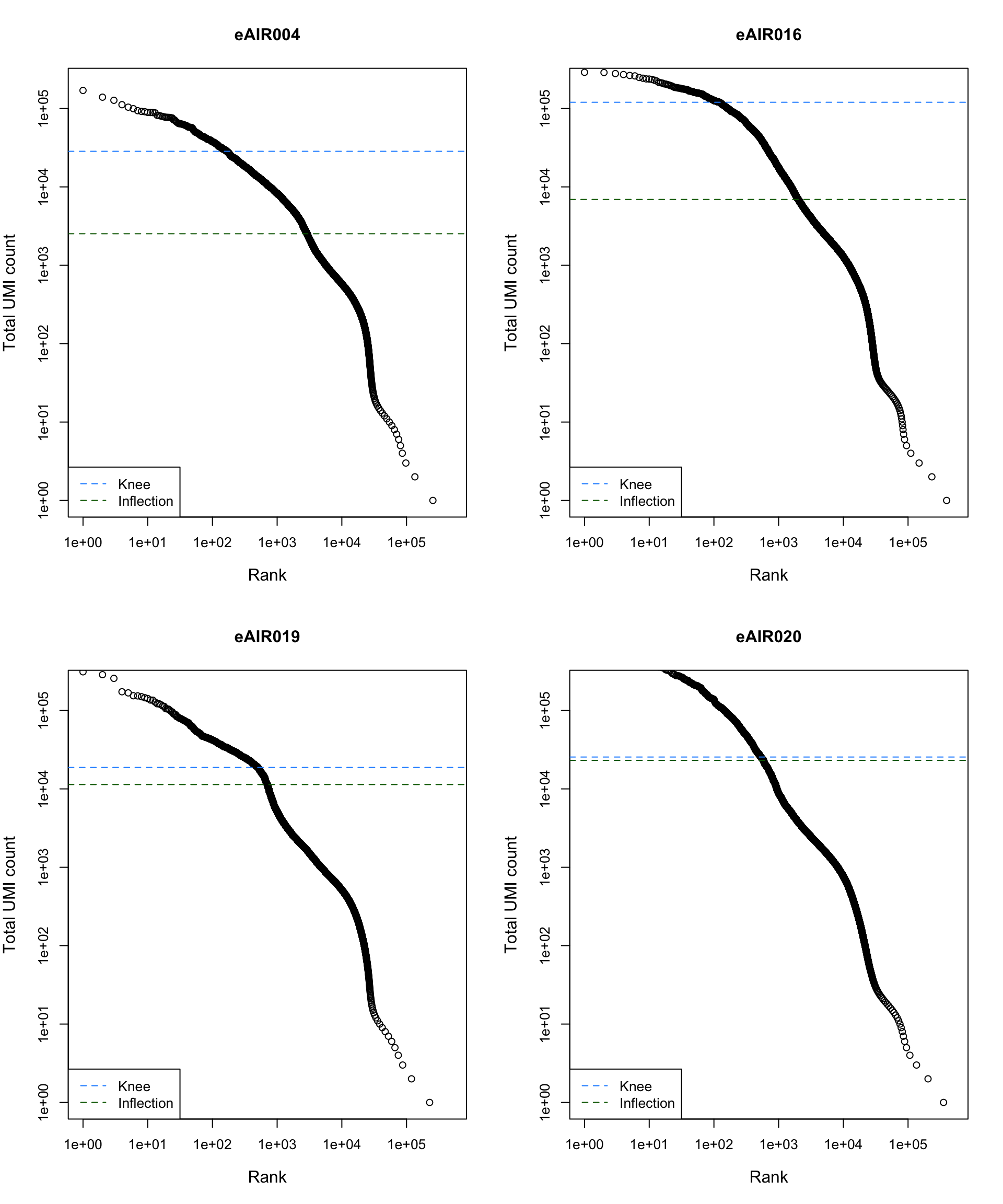
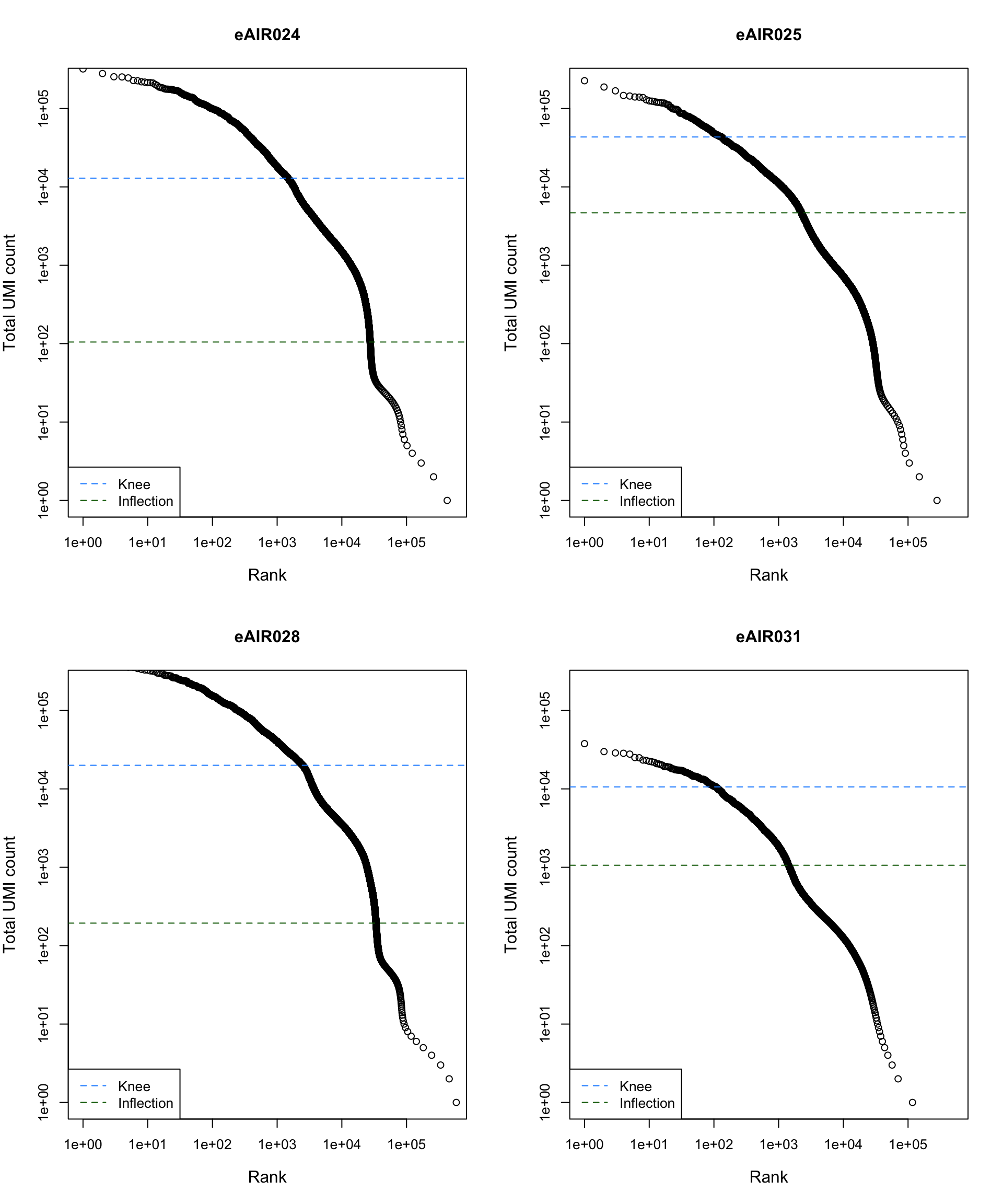
G000231_Neeland_batch3 - Adenoids
[1] “G000231_Neeland_batch3”
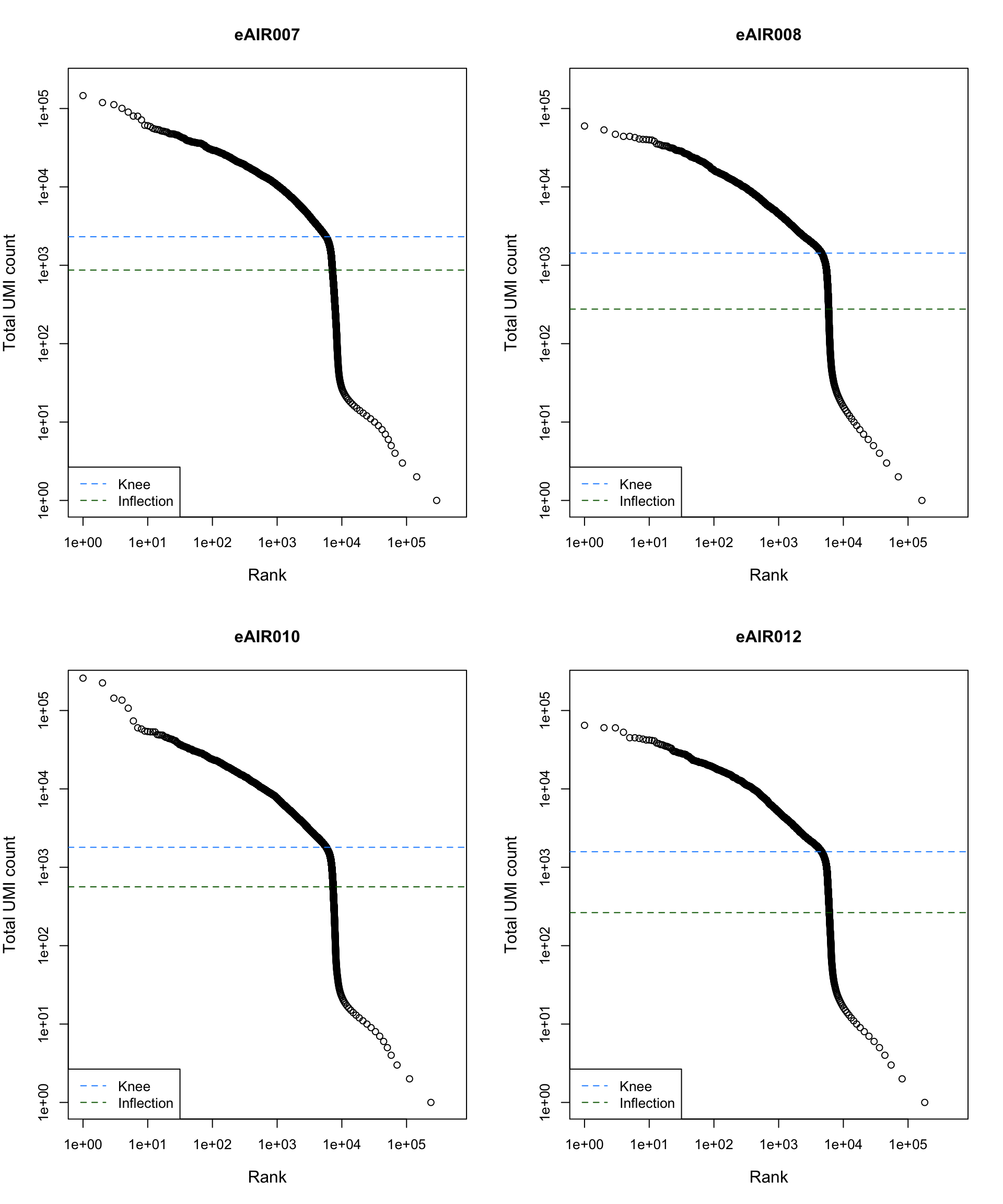
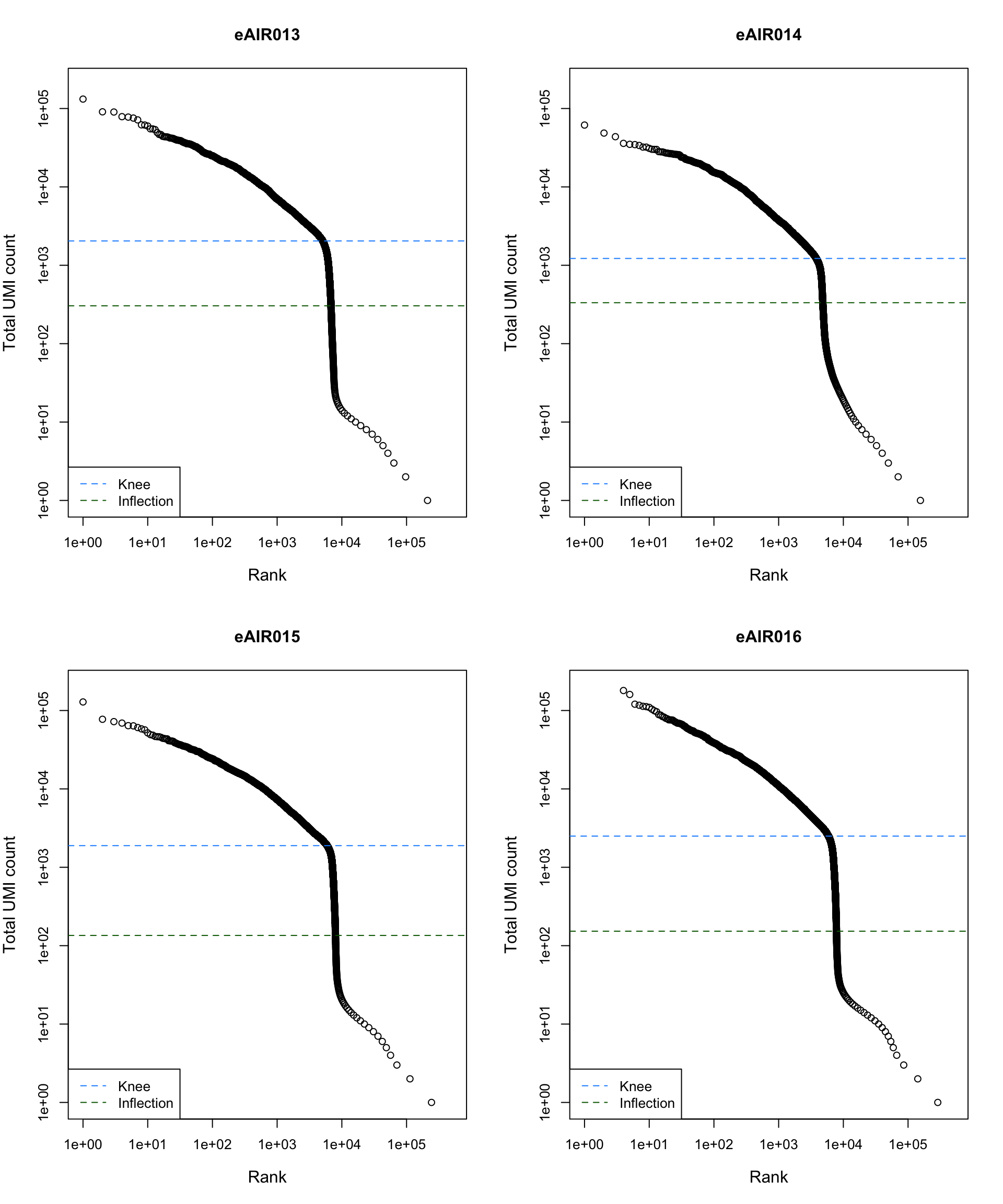
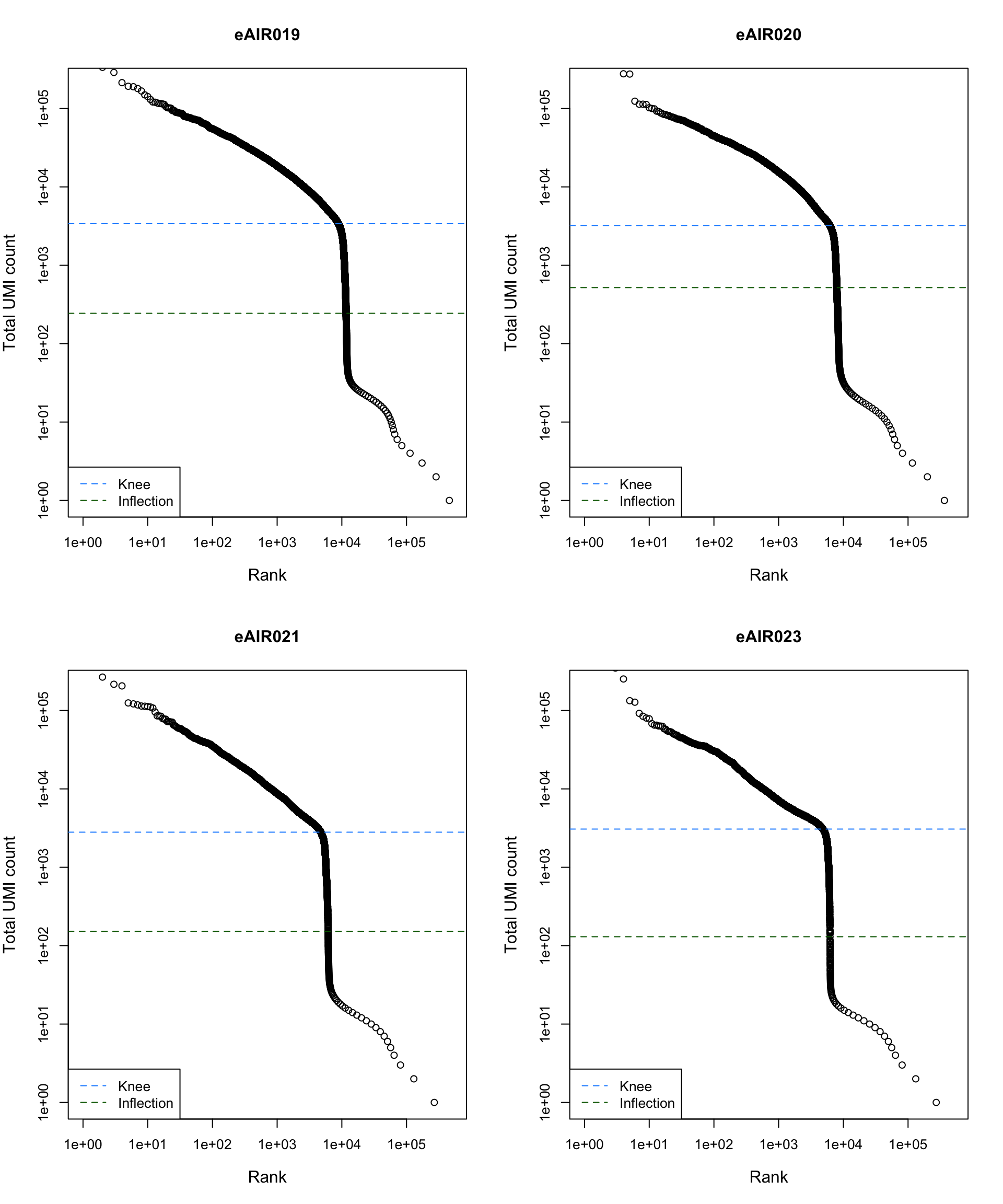
G000231_Neeland_batch4 - Bronchial_brushings
[1] “G000231_Neeland_batch4”
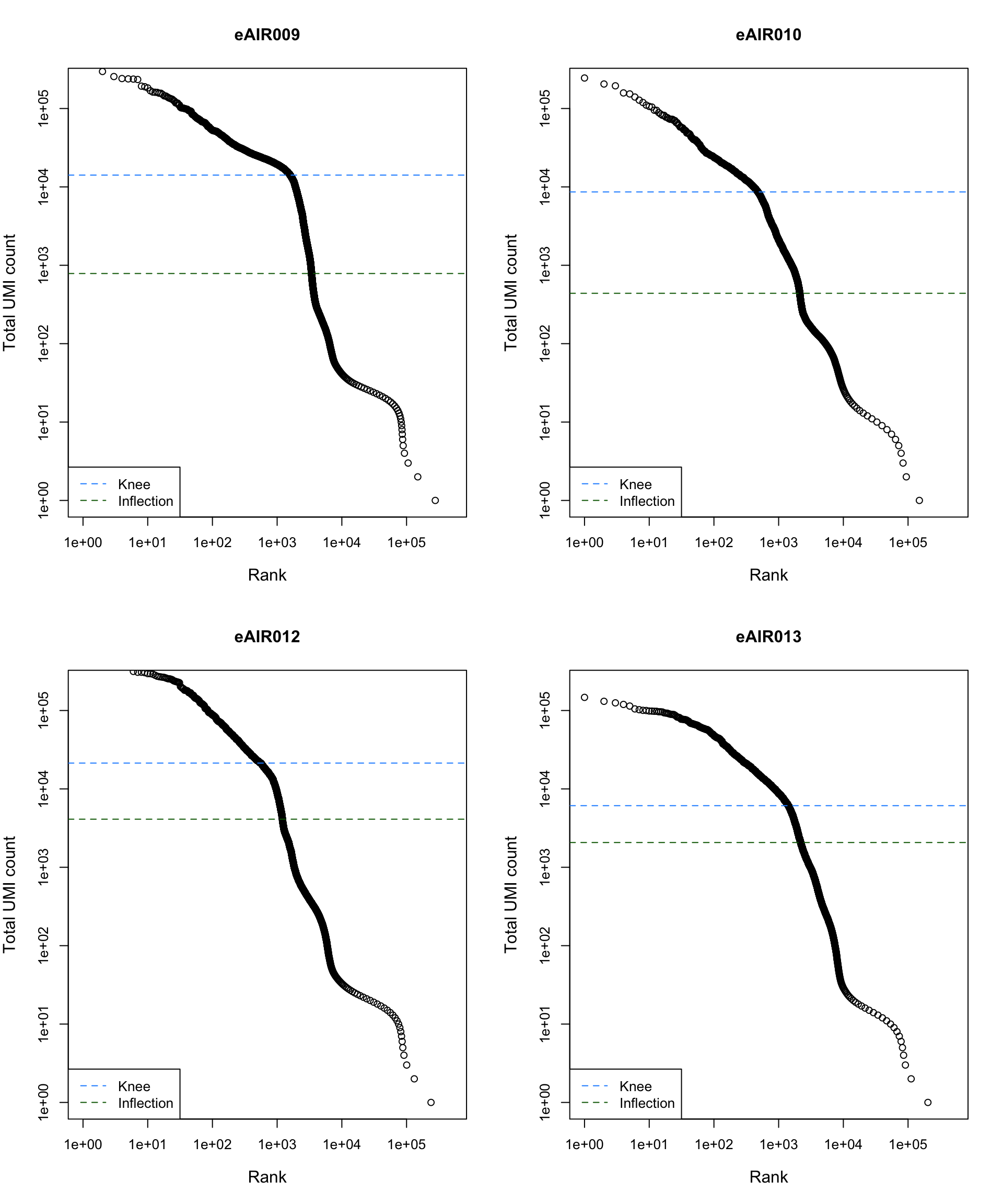
G000231_Neeland_batch5 - Nasal_brushings_2
[1] “G000231_Neeland_batch5”
# Reset the par settings to default after plotting
par(mfrow = c(1, 1))Session info
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_AU.UTF-8/en_AU.UTF-8/en_AU.UTF-8/C/en_AU.UTF-8/en_AU.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] kableExtra_1.3.4 DropletUtils_1.20.0
[3] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2
[5] Biobase_2.60.0 GenomicRanges_1.52.0
[7] GenomeInfoDb_1.36.1 IRanges_2.34.1
[9] S4Vectors_0.38.1 BiocGenerics_0.46.0
[11] MatrixGenerics_1.12.2 matrixStats_1.0.0
[13] glue_1.6.2 here_1.0.1
[15] lubridate_1.9.2 forcats_1.0.0
[17] stringr_1.5.0 dplyr_1.1.2
[19] purrr_1.0.1 readr_2.1.4
[21] tidyr_1.3.0 tibble_3.2.1
[23] ggplot2_3.4.2 tidyverse_2.0.0
[25] BiocStyle_2.28.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 rlang_1.1.1
[3] magrittr_2.0.3 git2r_0.32.0
[5] compiler_4.3.1 getPass_0.2-2
[7] DelayedMatrixStats_1.22.1 systemfonts_1.0.4
[9] callr_3.7.3 vctrs_0.6.3
[11] rvest_1.0.3 pkgconfig_2.0.3
[13] crayon_1.5.2 fastmap_1.1.1
[15] XVector_0.40.0 scuttle_1.10.1
[17] utf8_1.2.3 promises_1.2.0.1
[19] rmarkdown_2.23 tzdb_0.4.0
[21] ps_1.7.5 xfun_0.39
[23] zlibbioc_1.46.0 cachem_1.0.8
[25] beachmat_2.16.0 jsonlite_1.8.7
[27] highr_0.10 later_1.3.1
[29] rhdf5filters_1.12.1 DelayedArray_0.26.6
[31] Rhdf5lib_1.22.0 BiocParallel_1.34.2
[33] parallel_4.3.1 R6_2.5.1
[35] bslib_0.5.0 stringi_1.7.12
[37] limma_3.56.2 jquerylib_0.1.4
[39] Rcpp_1.0.11 knitr_1.43
[41] R.utils_2.12.2 httpuv_1.6.11
[43] Matrix_1.6-0 timechange_0.2.0
[45] tidyselect_1.2.0 rstudioapi_0.15.0
[47] yaml_2.3.7 codetools_0.2-19
[49] processx_3.8.2 lattice_0.21-8
[51] withr_2.5.0 evaluate_0.21
[53] xml2_1.3.5 pillar_1.9.0
[55] BiocManager_1.30.21.1 whisker_0.4.1
[57] generics_0.1.3 rprojroot_2.0.3
[59] RCurl_1.98-1.12 hms_1.1.3
[61] sparseMatrixStats_1.12.2 munsell_0.5.0
[63] scales_1.2.1 tools_4.3.1
[65] webshot_0.5.5 locfit_1.5-9.8
[67] fs_1.6.3 rhdf5_2.44.0
[69] grid_4.3.1 edgeR_3.42.4
[71] colorspace_2.1-0 GenomeInfoDbData_1.2.10
[73] HDF5Array_1.28.1 cli_3.6.1
[75] fansi_1.0.4 viridisLite_0.4.2
[77] S4Arrays_1.0.4 svglite_2.1.1
[79] gtable_0.3.3 R.methodsS3_1.8.2
[81] sass_0.4.7 digest_0.6.33
[83] dqrng_0.3.0 htmltools_0.5.5
[85] R.oo_1.25.0 lifecycle_1.0.3
[87] httr_1.4.6
sessionInfo()R version 4.3.1 (2023-06-16)
Platform: x86_64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.5
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_AU.UTF-8/en_AU.UTF-8/en_AU.UTF-8/C/en_AU.UTF-8/en_AU.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] kableExtra_1.3.4 DropletUtils_1.20.0
[3] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2
[5] Biobase_2.60.0 GenomicRanges_1.52.0
[7] GenomeInfoDb_1.36.1 IRanges_2.34.1
[9] S4Vectors_0.38.1 BiocGenerics_0.46.0
[11] MatrixGenerics_1.12.2 matrixStats_1.0.0
[13] glue_1.6.2 here_1.0.1
[15] lubridate_1.9.2 forcats_1.0.0
[17] stringr_1.5.0 dplyr_1.1.2
[19] purrr_1.0.1 readr_2.1.4
[21] tidyr_1.3.0 tibble_3.2.1
[23] ggplot2_3.4.2 tidyverse_2.0.0
[25] BiocStyle_2.28.0 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 rlang_1.1.1
[3] magrittr_2.0.3 git2r_0.32.0
[5] compiler_4.3.1 getPass_0.2-2
[7] DelayedMatrixStats_1.22.1 systemfonts_1.0.4
[9] callr_3.7.3 vctrs_0.6.3
[11] rvest_1.0.3 pkgconfig_2.0.3
[13] crayon_1.5.2 fastmap_1.1.1
[15] XVector_0.40.0 scuttle_1.10.1
[17] utf8_1.2.3 promises_1.2.0.1
[19] rmarkdown_2.23 tzdb_0.4.0
[21] ps_1.7.5 xfun_0.39
[23] zlibbioc_1.46.0 cachem_1.0.8
[25] beachmat_2.16.0 jsonlite_1.8.7
[27] highr_0.10 later_1.3.1
[29] rhdf5filters_1.12.1 DelayedArray_0.26.6
[31] Rhdf5lib_1.22.0 BiocParallel_1.34.2
[33] parallel_4.3.1 R6_2.5.1
[35] bslib_0.5.0 stringi_1.7.12
[37] limma_3.56.2 jquerylib_0.1.4
[39] Rcpp_1.0.11 knitr_1.43
[41] R.utils_2.12.2 httpuv_1.6.11
[43] Matrix_1.6-0 timechange_0.2.0
[45] tidyselect_1.2.0 rstudioapi_0.15.0
[47] yaml_2.3.7 codetools_0.2-19
[49] processx_3.8.2 lattice_0.21-8
[51] withr_2.5.0 evaluate_0.21
[53] xml2_1.3.5 pillar_1.9.0
[55] BiocManager_1.30.21.1 whisker_0.4.1
[57] generics_0.1.3 rprojroot_2.0.3
[59] RCurl_1.98-1.12 hms_1.1.3
[61] sparseMatrixStats_1.12.2 munsell_0.5.0
[63] scales_1.2.1 tools_4.3.1
[65] webshot_0.5.5 locfit_1.5-9.8
[67] fs_1.6.3 rhdf5_2.44.0
[69] grid_4.3.1 edgeR_3.42.4
[71] colorspace_2.1-0 GenomeInfoDbData_1.2.10
[73] HDF5Array_1.28.1 cli_3.6.1
[75] fansi_1.0.4 viridisLite_0.4.2
[77] S4Arrays_1.0.4 svglite_2.1.1
[79] gtable_0.3.3 R.methodsS3_1.8.2
[81] sass_0.4.7 digest_0.6.33
[83] dqrng_0.3.0 htmltools_0.5.5
[85] R.oo_1.25.0 lifecycle_1.0.3
[87] httr_1.4.6