BAL: WithoutdecontX
Clustering and Marker gene analysis without running DecontX(ambient removal)
Gunjan Dixit
October 11, 2024
Last updated: 2024-10-11
Checks: 6 1
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 676bf00. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/Cell_labels_Mel_v3/
Ignored: data/RDS/
Ignored: output/.DS_Store
Ignored: output/CSV/.DS_Store
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Ignored: output/G000231_Neeland_batch9_1/
Ignored: output/RDS/
Ignored: output/plots/
Untracked files:
Untracked: Adenoids_Bcell_subset_proportions_Age.pdf
Untracked: Adenoids_Tcell_subset_proportions_Age.pdf
Untracked: Adenoids_cell_type_proportions_Age.pdf
Untracked: Age_proportions_Adenoids.pdf
Untracked: Age_proportions_Bronchial_brushings.pdf
Untracked: Age_proportions_Nasal_brushings.pdf
Untracked: Age_proportions_Tonsils.pdf
Untracked: BAL_Tcell_propeller.xlsx
Untracked: BAL_propeller.xlsx
Untracked: BB_Tcell_propeller.xlsx
Untracked: BB_propeller.xlsx
Untracked: NB_Tcell_propeller.xlsx
Untracked: NB_propeller.csv
Untracked: NB_propeller.pdf
Untracked: NB_propeller.xlsx
Untracked: Tonsils_cell_type_proportions.jpg
Untracked: Tonsils_cell_type_proportions.pdf
Untracked: Tonsils_cell_type_proportions.png
Untracked: Tonsils_cell_type_proportions_Age.pdf
Untracked: analysis/03_Batch_Integration.Rmd
Untracked: analysis/Age_proportions.Rmd
Untracked: analysis/Age_proportions_AllBatches.Rmd
Untracked: analysis/Batch_Integration_&_Downstream_analysis.Rmd
Untracked: analysis/Batch_correction_&_Downstream.Rmd
Untracked: analysis/Cell_cycle_regression.Rmd
Untracked: analysis/Master_metadata.Rmd
Untracked: analysis/Preprocessing_Batch1_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch2_Tonsils.Rmd
Untracked: analysis/Preprocessing_Batch3_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch4_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch5_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch6_BAL.Rmd
Untracked: analysis/Preprocessing_Batch7_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch8_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch9_Tonsils.Rmd
Untracked: analysis/TonsilsVsAdenoids.Rmd
Untracked: analysis/cell_cycle_regression.R
Untracked: analysis/test.Rmd
Untracked: analysis/testing_age_all.Rmd
Untracked: cell_proportions_overview.png
Untracked: cell_type_proportions.pdf
Untracked: cell_type_proportions_enhanced.pdf
Untracked: cell_type_proportions_individual.pdf
Untracked: color_palette.rds
Untracked: color_palette_v2_level2.rds
Untracked: combined_metadata.rds
Untracked: data/Cell_labels_Mel/
Untracked: data/Cell_labels_Mel_v2/
Untracked: data/Cell_labels_modified_Gunjan/
Untracked: data/Hs.c2.cp.reactome.v7.1.entrez.rds
Untracked: data/Raw_feature_bc_matrix/
Untracked: data/celltypes_Mel_GD_v3.xlsx
Untracked: data/celltypes_Mel_GD_v4_no_dups.xlsx
Untracked: data/celltypes_Mel_modified.xlsx
Untracked: data/celltypes_Mel_v2.csv
Untracked: data/celltypes_Mel_v2.xlsx
Untracked: data/celltypes_Mel_v2_MN.xlsx
Untracked: data/celltypes_for_mel_MN.xlsx
Untracked: data/earlyAIR_sample_sheets_combined.xlsx
Untracked: output/CSV/All_tissues.propeller.xlsx
Untracked: output/CSV/Bronchial_brushings/
Untracked: output/CSV/Bronchial_brushings_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/
Untracked: output/CSV/G000231_Neeland_Adenoids.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Bronchial_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Nasal_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Tonsils.propeller.xlsx
Untracked: output/CSV/Nasal_brushings/
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Modified: analysis/00_AllBatches_overview.Rmd
Modified: analysis/01_QC_emptyDrops.Rmd
Modified: analysis/02_QC_exploratoryPlots.Rmd
Modified: analysis/Adenoids.Rmd
Modified: analysis/Age_modeling.Rmd
Modified: analysis/AllBatches_QCExploratory.Rmd
Modified: analysis/BAL.Rmd
Modified: analysis/BAL_without_DecontX.Rmd
Modified: analysis/Bronchial_brushings.Rmd
Modified: analysis/Nasal_brushings.Rmd
Modified: analysis/Subclustering_Nasal_brushings.Rmd
Modified: analysis/Tonsils.Rmd
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c9.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c9.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/BAL_without_DecontX.Rmd)
and HTML (docs/BAL_without_DecontX.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 0f07f72 | Gunjan Dixit | 2024-10-07 | Added BAL subclustering without DecontX |
| html | 0f07f72 | Gunjan Dixit | 2024-10-07 | Added BAL subclustering without DecontX |
| Rmd | 781f596 | Gunjan Dixit | 2024-10-01 | Added BAL without DecontX analysis |
| html | 781f596 | Gunjan Dixit | 2024-10-01 | Added BAL without DecontX analysis |
Introduction
This RMarkdown file loads and analyzes the Seurat object for BAL (Batch6) without running decontX (ambient removal) and compares the findings or the cells that change with/without ambient removal.
Load libraries
suppressPackageStartupMessages({
library(BiocStyle)
library(tidyverse)
library(here)
library(glue)
library(dplyr)
library(Seurat)
library(clustree)
library(kableExtra)
library(RColorBrewer)
library(data.table)
library(ggplot2)
library(readr)
library(patchwork)
library(limma)
library(edgeR)
library(speckle)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(readxl)
})Load Input data
For BAL, we had just one batch- Batch6.
tissue <- "BAL"Clustering
Clustering is done on the “pca” reduction at resolutions ranging from 0-1.
out1 <- here("output",
"RDS", "AllBatches_Clustering_SEUs",
paste0("G000231_Neeland_",tissue,"_without_decontX.Clusters.SEU.rds"))
#dir.create(out1)
resolutions <- seq(0.1, 1, by = 0.1)
if (!file.exists(out1)) {
seu_obj <- FindNeighbors(seu_obj, reduction = "pca", dims = 1:30)
seu_obj <- FindClusters(seu_obj, resolution = seq(0.1, 1, by = 0.1), algorithm = 3)
saveRDS(seu_obj, file = out1)
} else {
seu_obj <- readRDS(out1)
}The clustree function is used to visualize the
clustering at different resolutions to identify the most optimum
resolution.
clustree(seu_obj, prefix = "RNA_snn_res.")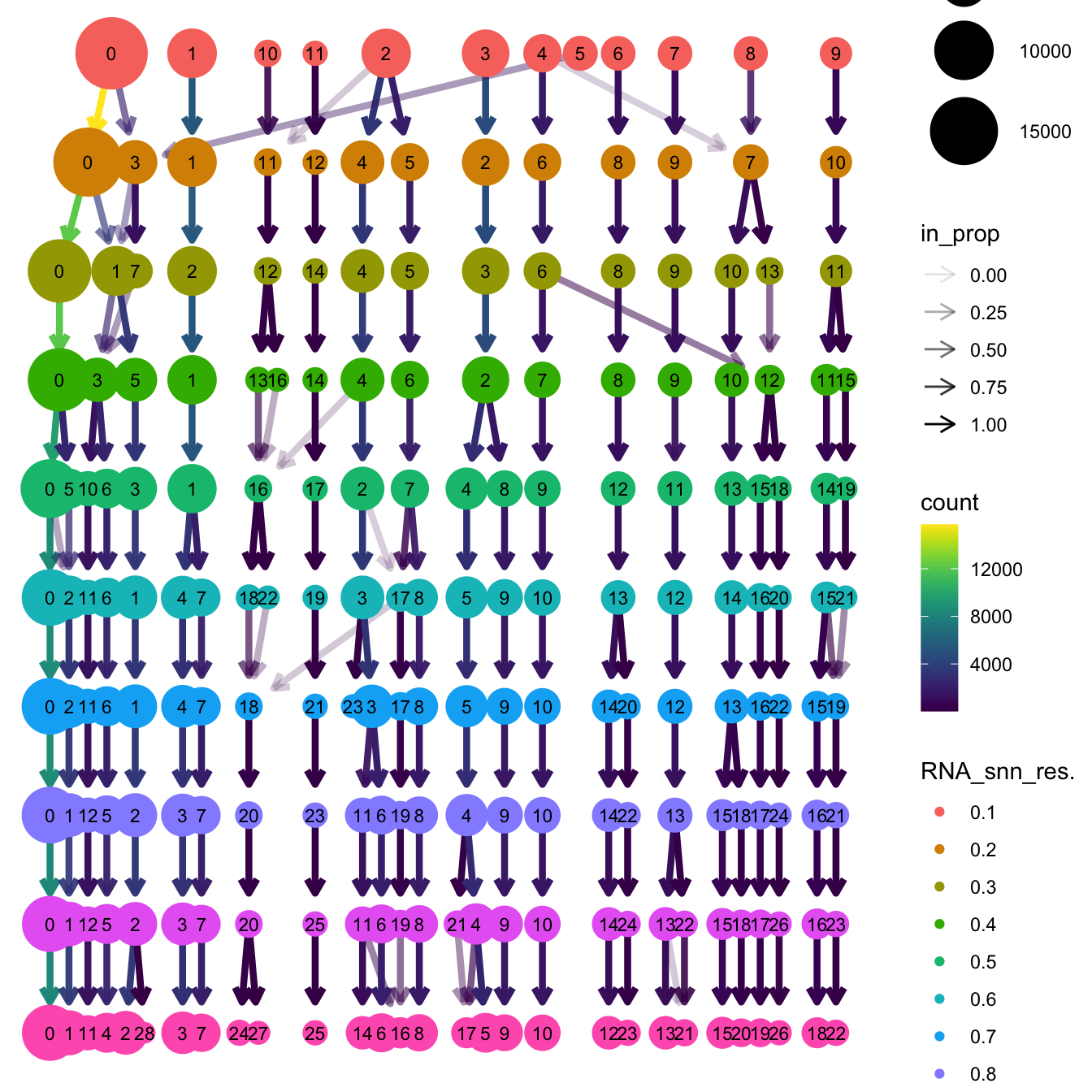
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Based on the clustering tree, we chose an intermediate/optimum resolution where the clustering results are the most stable, with the least amount of shuffling cells.
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(seu_obj$RNA_snn_res.0.4)
seu_obj$RNA_snn_res.0.4 <- factor(seu_obj$RNA_snn_res.0.4, levels = seq(0,n-1))
seu_obj$seurat_clusters <- NULL
seu_obj$cluster <- seu_obj$RNA_snn_res.0.4
Idents(seu_obj) <- seu_obj$clusterUMAP after clustering
Defining colours for each cell-type to be consistent with other age-related/cell type composition plots.
my_colors <- c(
"B cells" = "steelblue",
"CD4 T cells" = "brown",
"Double negative T cells" = "gold",
"CD8 T cells" = "lightgreen",
"Pre B/T cells" = "orchid",
"Innate lymphoid cells" = "tan",
"Natural Killer cells" = "blueviolet",
"Macrophages" = "green4",
"Cycling T cells" = "turquoise",
"Dendritic cells" = "grey80",
"Gamma delta T cells" = "mediumvioletred",
"Epithelial lineage" = "darkorange",
"Granulocytes" = "olivedrab",
"Fibroblast lineage" = "lavender",
"None" = "white",
"Monocytes" = "peachpuff",
"Endothelial lineage" = "cadetblue",
"SMG duct" = "lightpink",
"Neuroendocrine" = "skyblue",
"Doublet query/Other" = "#d62728"
)UMAP displaying clusters at opt_res resolution and Broad
cell Labels Level 3.
p1 <- DimPlot(seu_obj, reduction = "umap", raster = FALSE ,repel = TRUE, label = TRUE,label.size = 3.5, group.by = opt_res) + NoLegend()
p2 <- DimPlot(seu_obj, reduction = "umap", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5, group.by = "Broad_cell_label_3") +
scale_colour_manual(values = my_colors) +
ggtitle(paste0(tissue, ": UMAP (Azimuth Labels)"))
p1 / p2 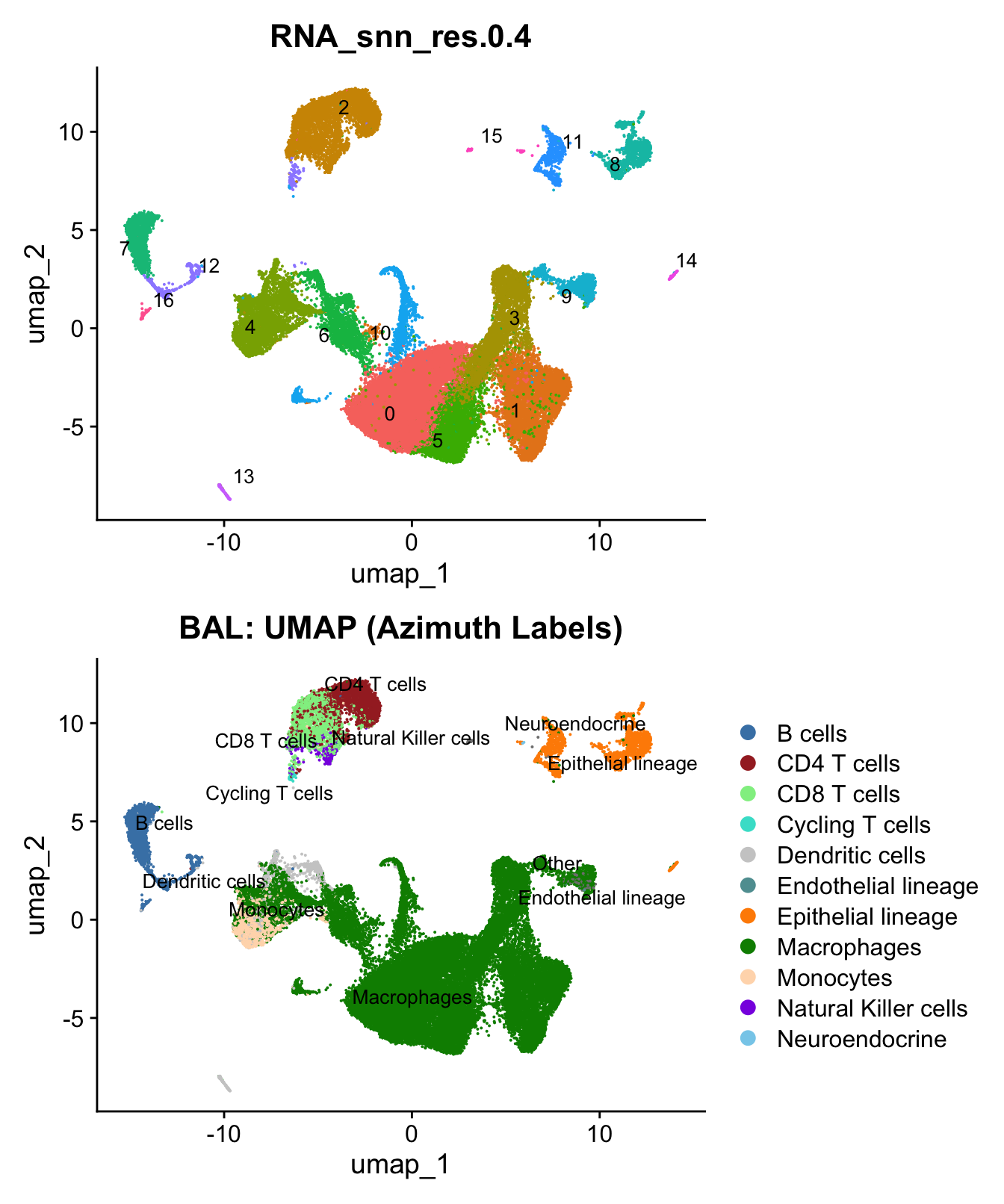
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Marker Gene Analysis
Here is the link to marker gene analysis of BAL (without ambient removal) BAL_withoutDecontX_res.0.4
seu_obj <- JoinLayers(seu_obj)
paed.markers <- FindAllMarkers(seu_obj, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Extracting top 5 genes per cluster for visualization. The ‘top5’ contains the top 5 genes with the highest weighted average avg_log2FC within each cluster and the ‘best.wilcox.gene.per.cluster’ contains the single best gene with the highest weighted average avg_log2FC for each cluster.
paed.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "THBS1" "DEFB1" "IL32" "NR1H3" "SOCS3" "TMEM74B" "VCAN"
[8] "CD79A" "CTXN1" "SLC11A1" "TK1" "SLPI" "ATP2A3" "PTPRS"
[15] "KRT13" "KIT" "JCHAIN" Marker gene expression in clusters
This heatmap depicts the expression of top five genes in each cluster.
DoHeatmap(seu_obj, features = top5$gene) + NoLegend()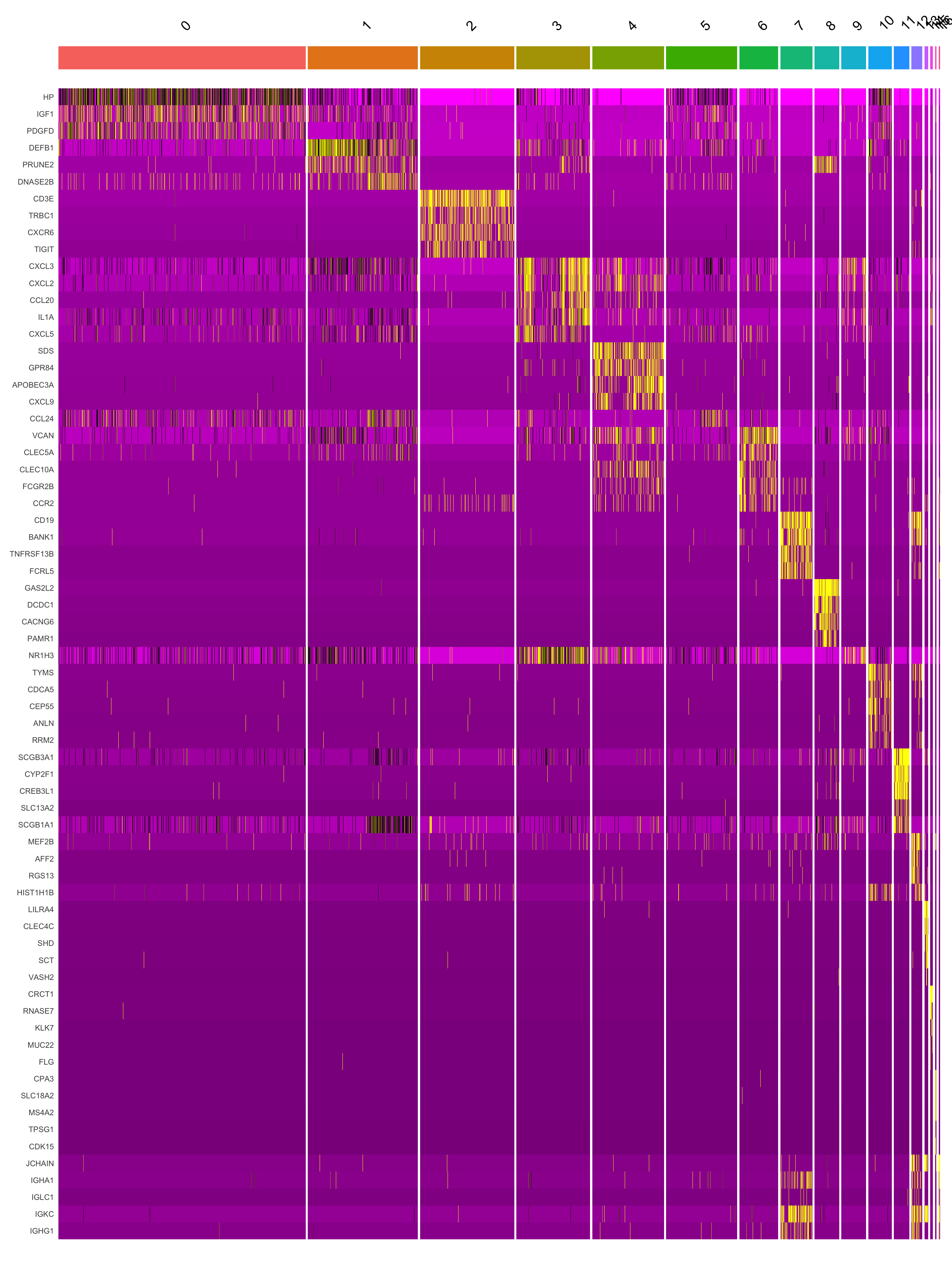
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Violin plot shows the expression of top marker gene per cluster.
VlnPlot(seu_obj, features=best.wilcox.gene.per.cluster, ncol = 2, raster = FALSE, pt.size = FALSE)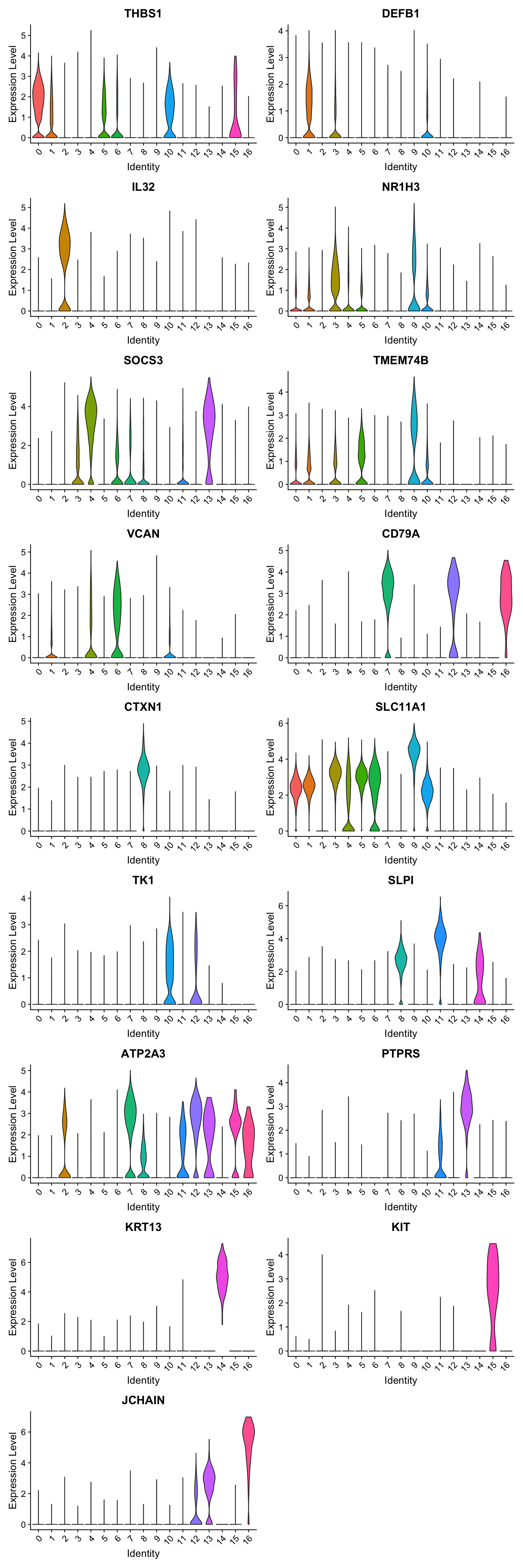
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(seu_obj,features=best.wilcox.gene.per.cluster, reduction = 'umap', raster = FALSE, ncol = 2)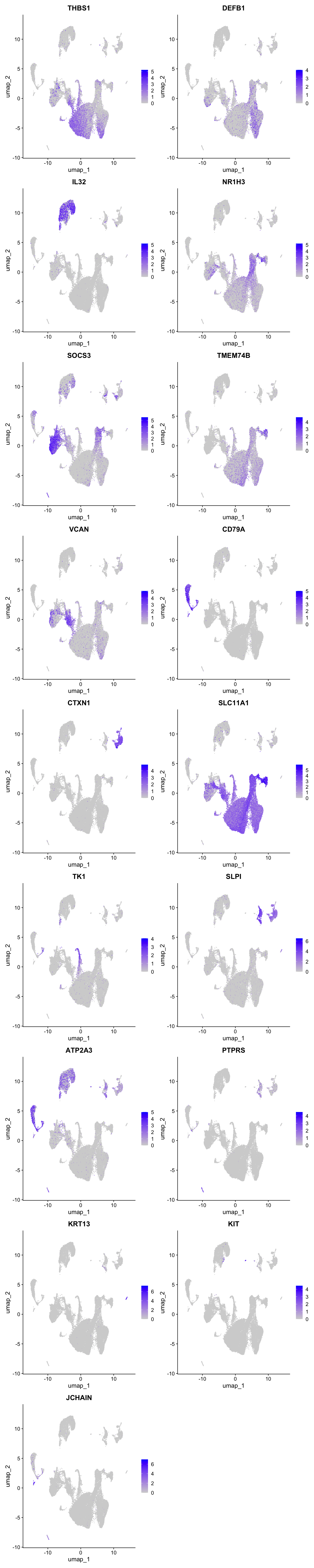
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(seu_obj,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.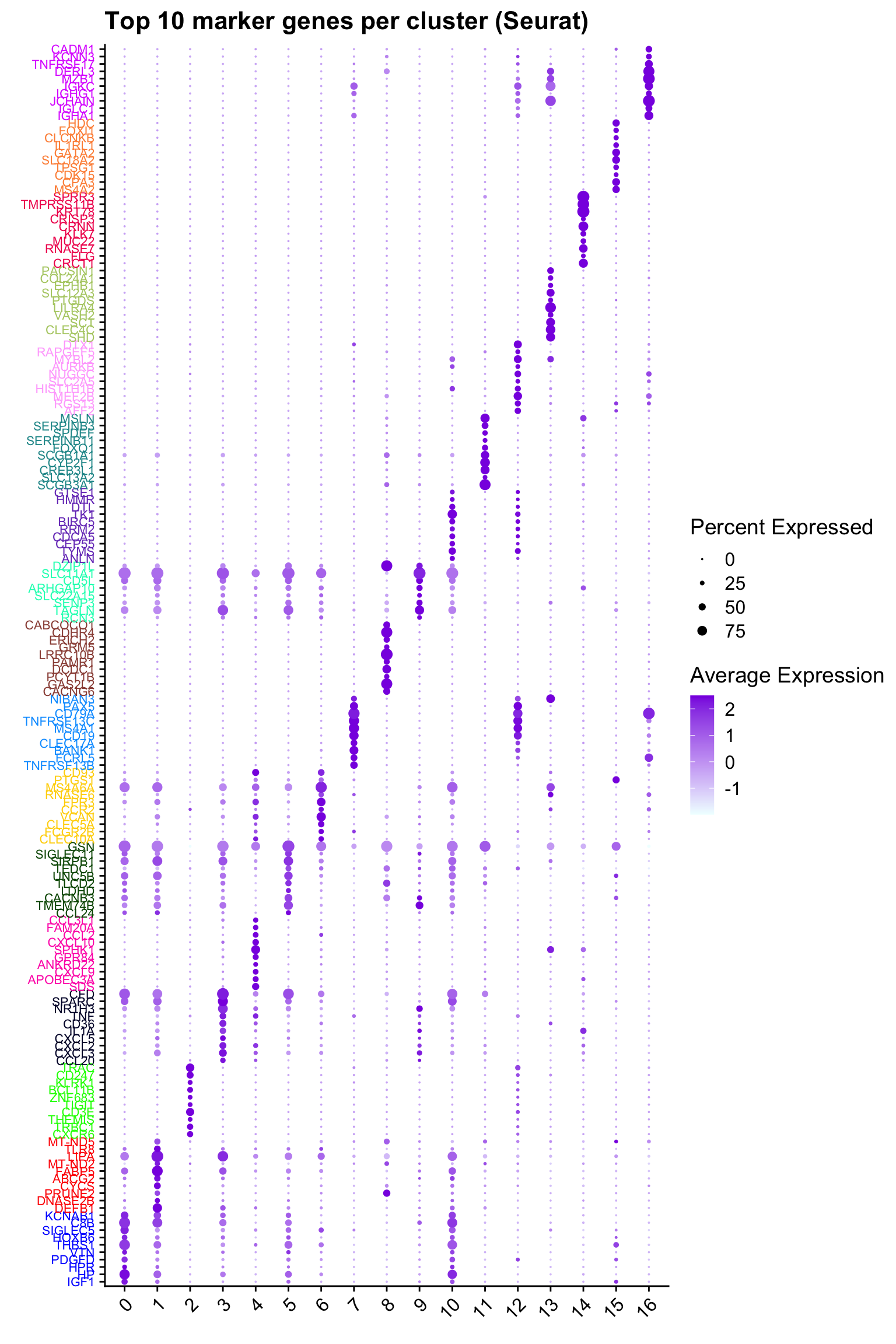
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
Extract markers for each cluster
This section extracts marker genes for each cluster and save them as a CSV file.
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_gene_clusters_withoutDecontX.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed.markers$cluster)) {
cluster_data <- paed.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
}Update Macro labels
cell_labels <- readxl::read_excel(here("data/Cell_labels_Mel_v2/earlyAIR_BAL_withoutDecontX_annotations_03.10.24_with broad label and flow label.xlsx"))
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
seu_obj <- RenameIdents(seu_obj, new_cluster_names)
seu_obj@meta.data$cell_labels <- Idents(seu_obj)
p3 <- DimPlot(seu_obj, reduction = "umap", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated cell types"))
p3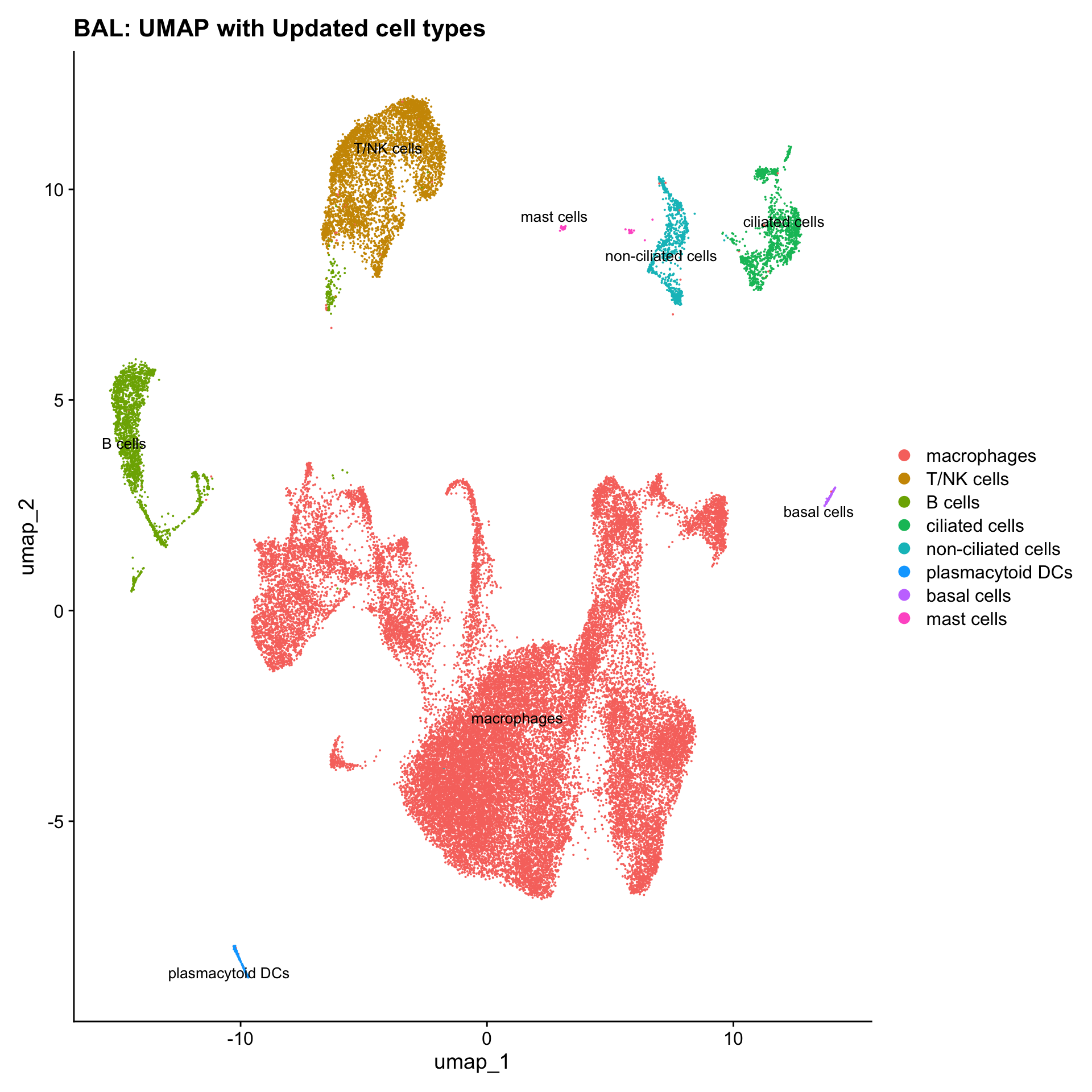
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
Reclustering Macro polulation
Here is the link to marker gene analysis of Macrophages in BAL (without ambient removal) BAL_macro_withoutDecontX_res.0.7
Clusters corresponding to macrophage cell type are-
idx <- which(Idents(seu_obj) %in% c("macrophages"))
paed_sub <- seu_obj[,idx]
paed_sub@meta.data$donor <- sub("_\\d+$", "", paed_sub@meta.data$donor_id)
paed_subAn object of class Seurat
17529 features across 32502 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
2 dimensional reductions calculated: pca, umappaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9687
Number of communities: 8
Elapsed time: 25 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9539
Number of communities: 10
Elapsed time: 26 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9398
Number of communities: 11
Elapsed time: 25 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9263
Number of communities: 12
Elapsed time: 24 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9162
Number of communities: 16
Elapsed time: 21 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9082
Number of communities: 18
Elapsed time: 21 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9012
Number of communities: 21
Elapsed time: 20 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8944
Number of communities: 21
Elapsed time: 20 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8877
Number of communities: 23
Elapsed time: 20 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32502
Number of edges: 1049780
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8815
Number of communities: 24
Elapsed time: 19 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)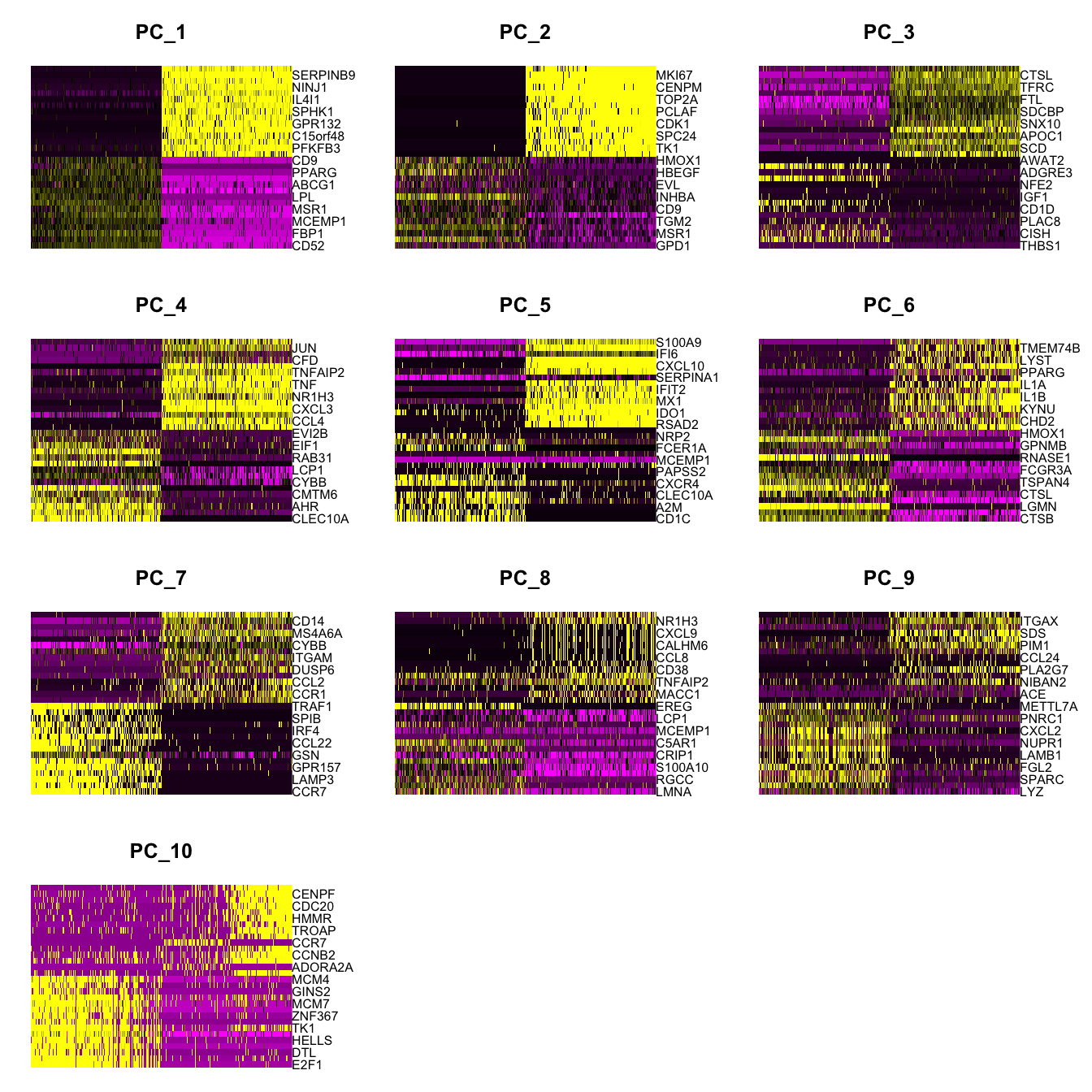
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
clustree(paed_sub, prefix = "RNA_snn_res.")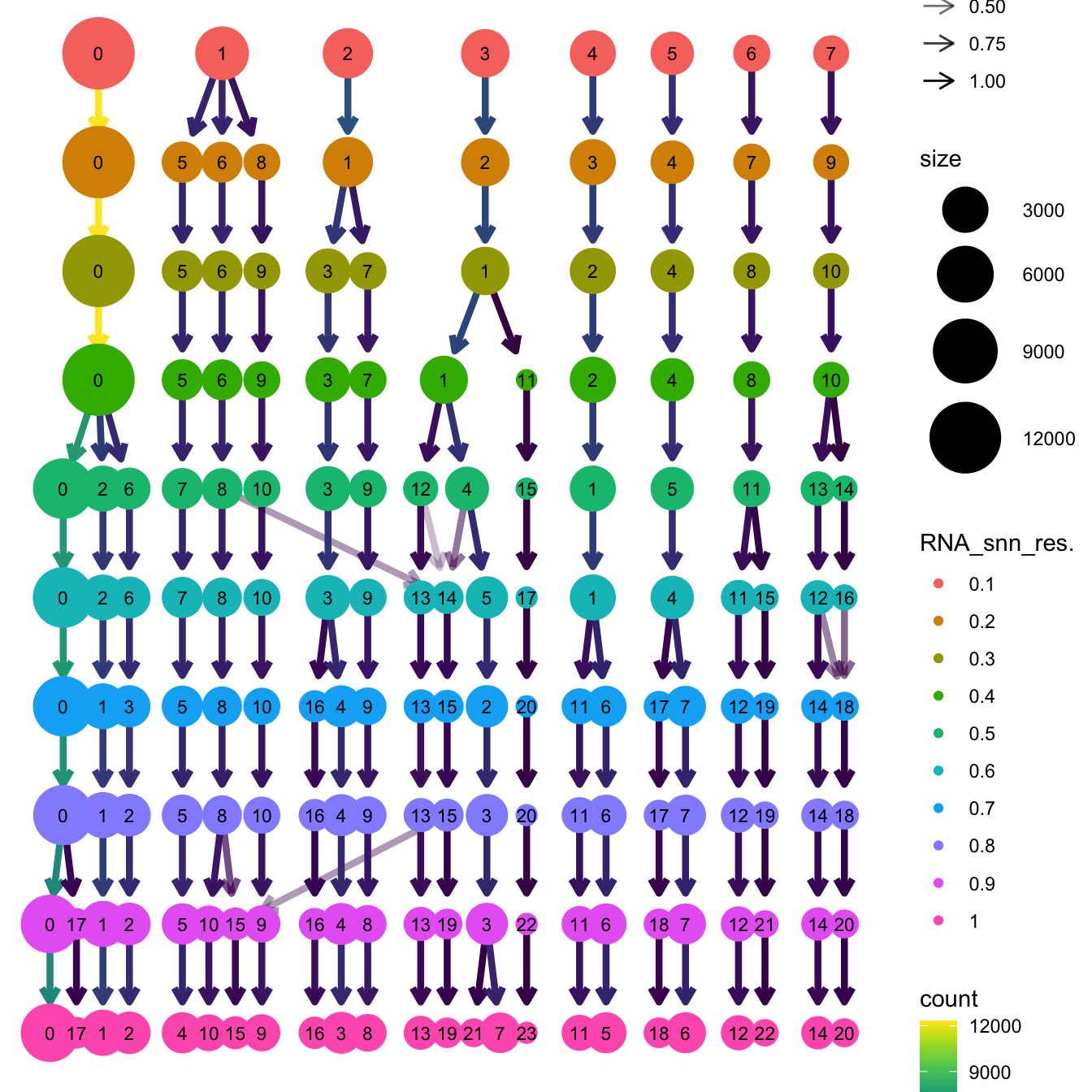
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
opt_res <- "RNA_snn_res.0.7"
n <- nlevels(paed_sub$RNA_snn_res.0.7)
paed_sub$RNA_snn_res.0.7 <- factor(paed_sub$RNA_snn_res.0.7, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
Idents(paed_sub) <- paed_sub$RNA_snn_res.0.7DimPlot(paed_sub, reduction = "umap.new", group.by = "RNA_snn_res.0.7", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )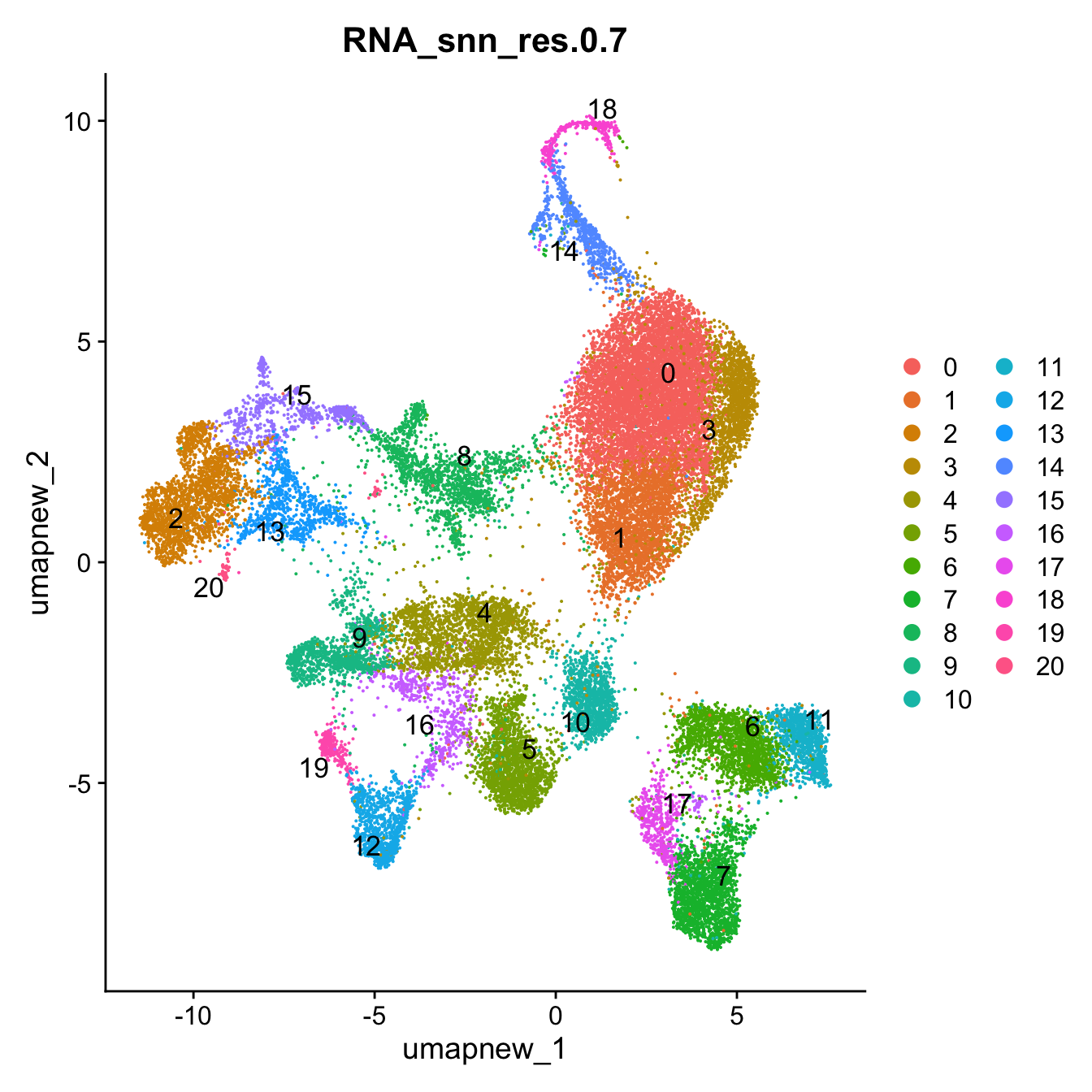
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_level_4", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )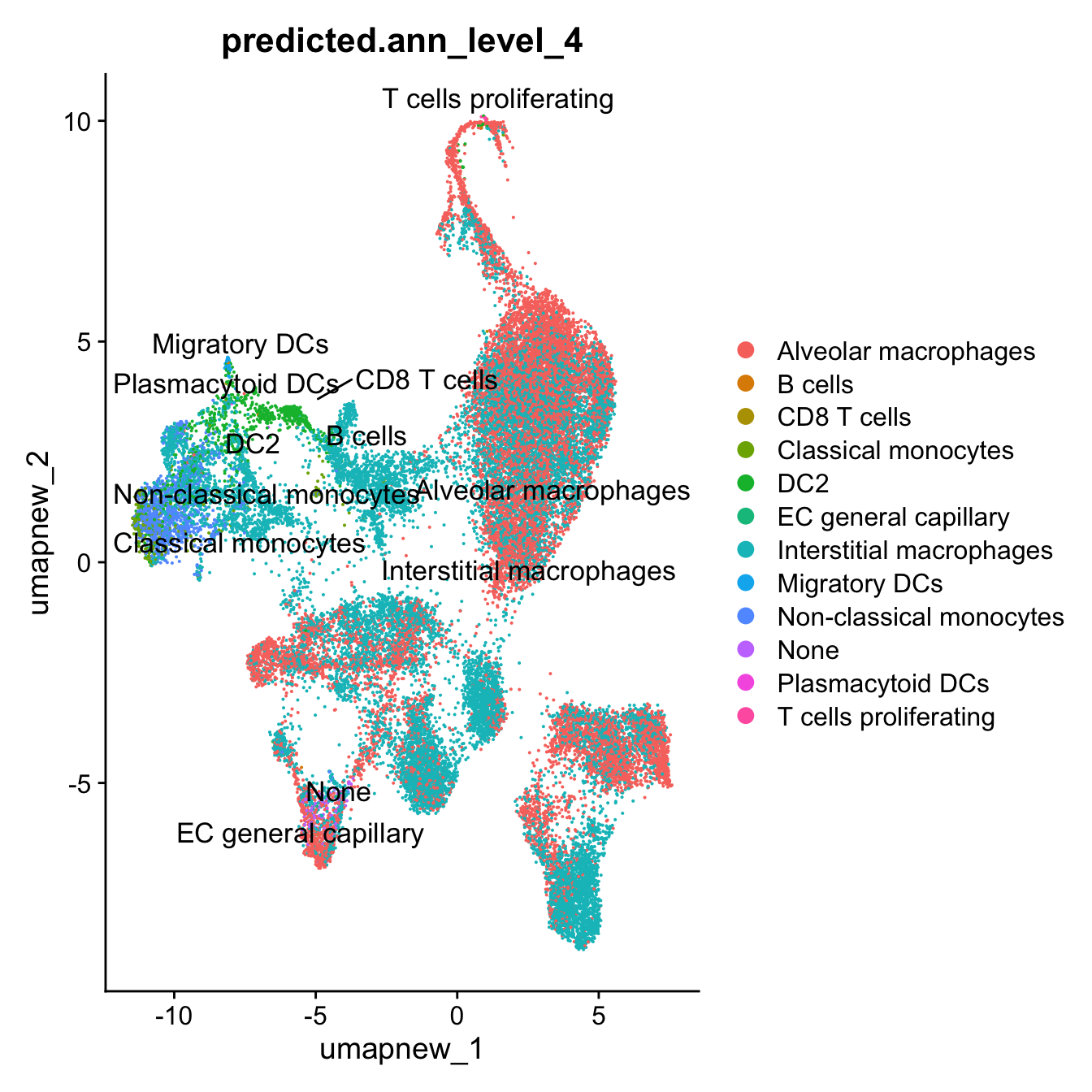
Azimuth Labels
## Finest level
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_finest_level", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) 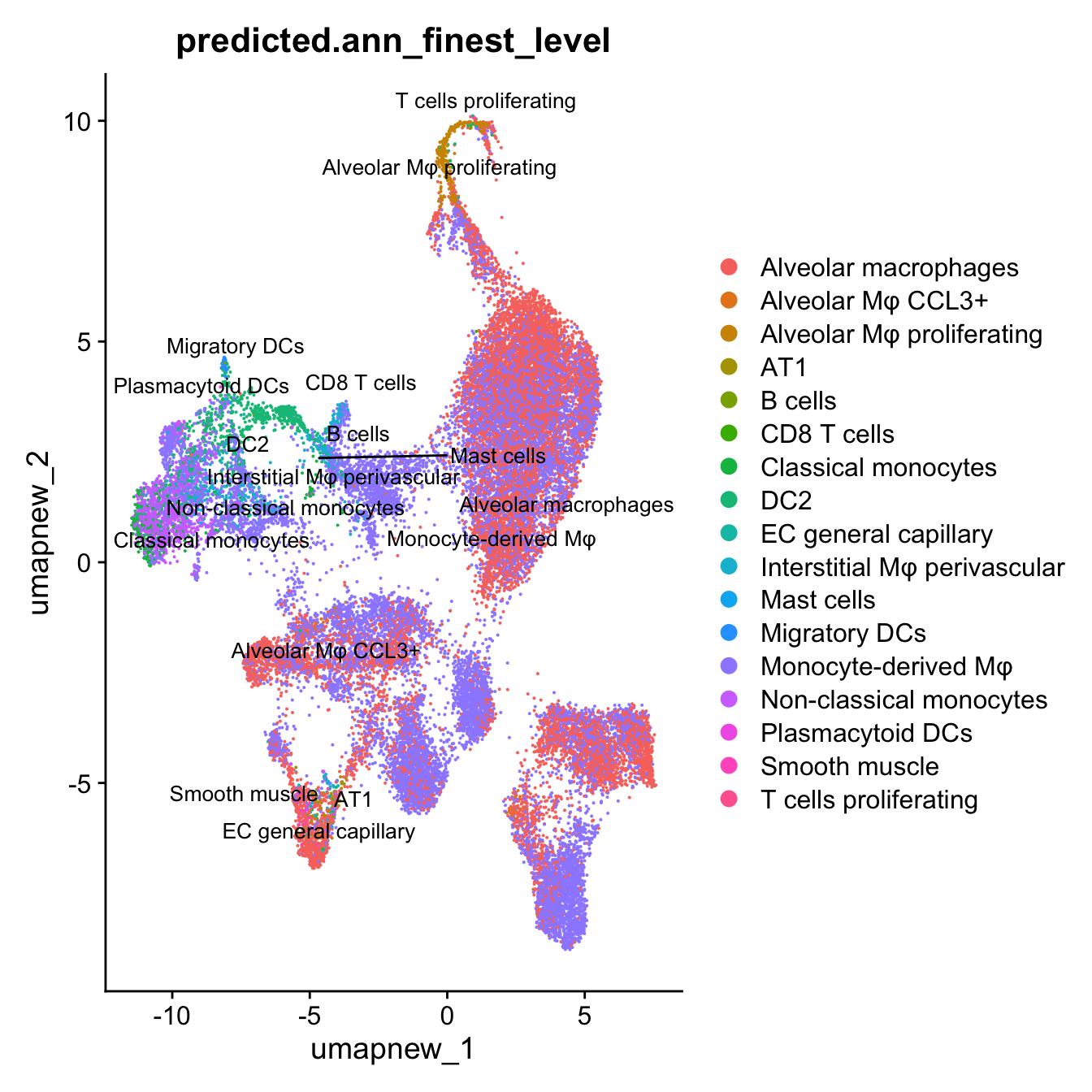
df_table <- as.data.frame(table(paed_sub$RNA_snn_res.0.7, paed_sub$predicted.ann_finest_level))
ggplot(df_table, aes(Var1, Freq, fill = Var2)) +
geom_bar(stat = "identity") +
labs(x = "RNA_snn_res.0.7", y = "Count", fill = "predicted ann_finest_level") +
theme_minimal() +
ggtitle("Stacked Bar Plot Azimuth predicted.ann_finest_level")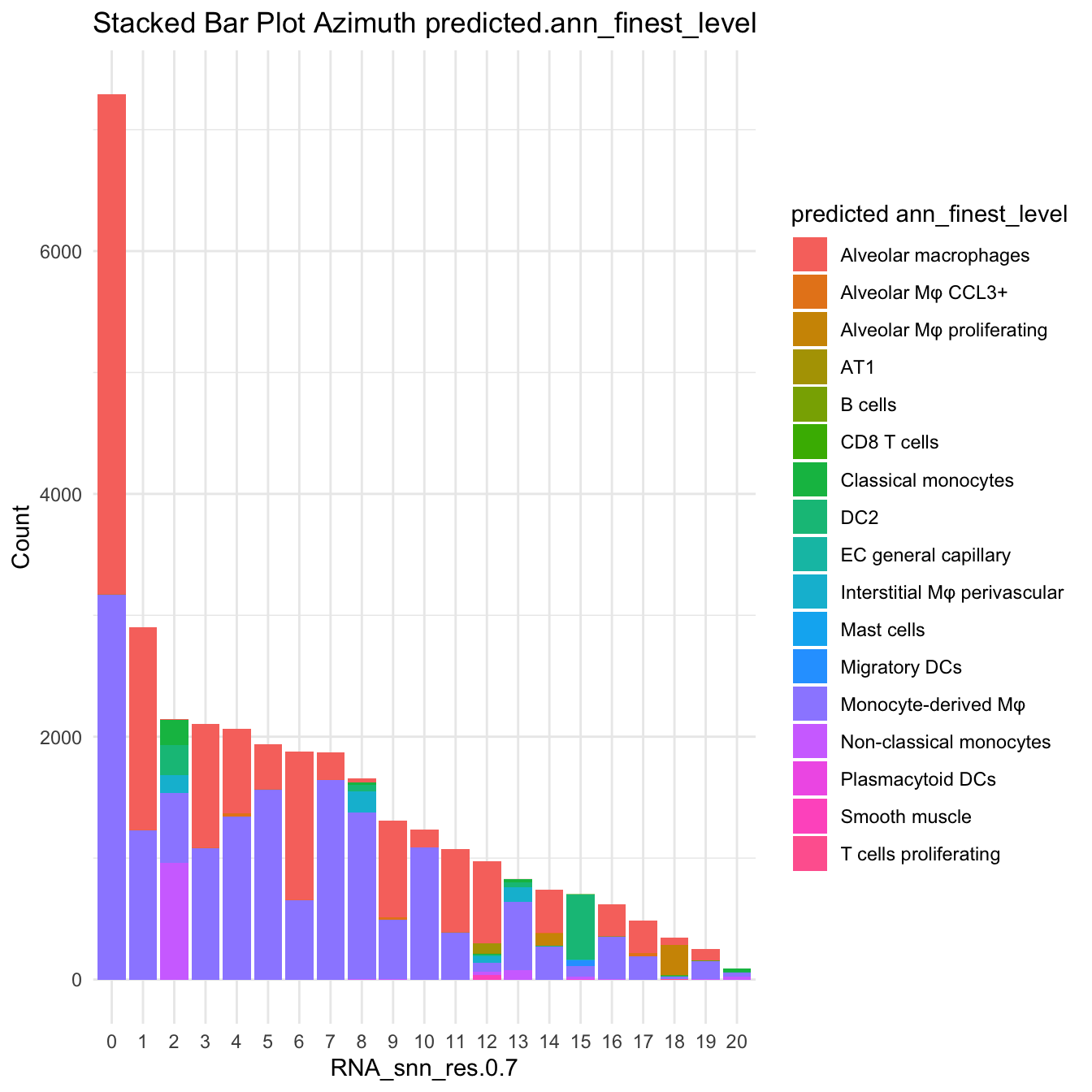
| Version | Author | Date |
|---|---|---|
| 781f596 | Gunjan Dixit | 2024-10-01 |
## Predicted_Level 5
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_level_4", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) 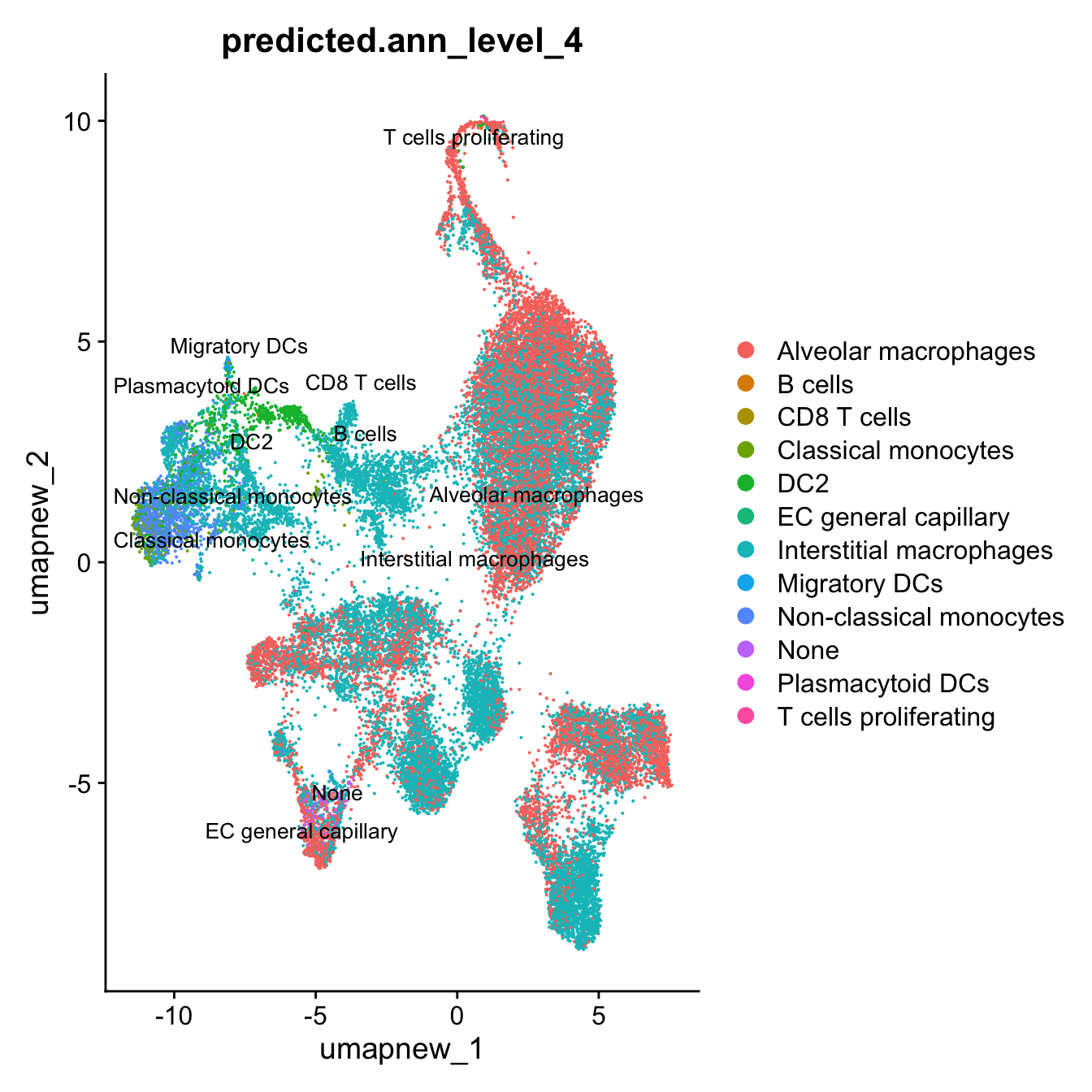
df_table <- as.data.frame(table(paed_sub$RNA_snn_res.0.7, paed_sub$predicted.ann_level_4))
ggplot(df_table, aes(Var1, Freq, fill = Var2)) +
geom_bar(stat = "identity") +
labs(x = "RNA_snn_res.0.7", y = "Count", fill = "predicted ann_level_4") +
theme_minimal() +
ggtitle("Stacked Bar Plot Azimuth label")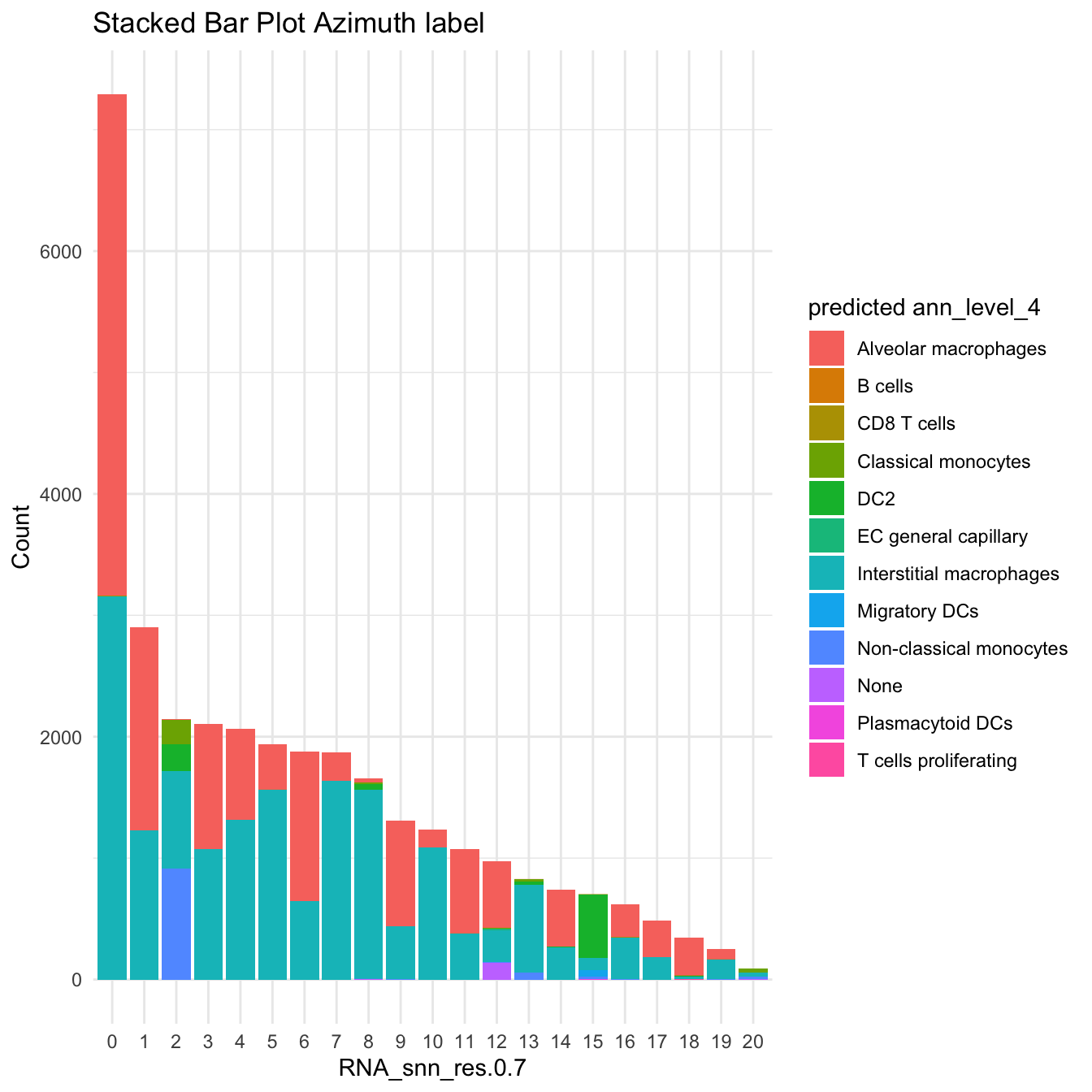
palette1 <- paletteer::paletteer_d("ggthemes::Classic_20")
palette2 <- paletteer::paletteer_d("Polychrome::light")
combined_palette <- unique(c(palette1, palette2))
p2 <- paed_sub@meta.data %>%
dplyr::select(!!sym(opt_res), donor) %>%
group_by(!!sym(opt_res), donor) %>%
summarise(num = n()) %>%
mutate(prop = num / sum(num)) %>%
ggplot(aes(x = !!sym(opt_res), y = prop * 100,
fill = donor)) +
geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90,
vjust = 0.5,
hjust = 1,
size = 8)) +
labs(y = "% Cells", fill = "donor") +
scale_fill_manual(values = combined_palette)`summarise()` has grouped output by 'RNA_snn_res.0.7'. You can override using
the `.groups` argument.# Combine the plots
p2 & theme( legend.text = element_text(size = 8),
legend.key.size = unit(3, "mm"))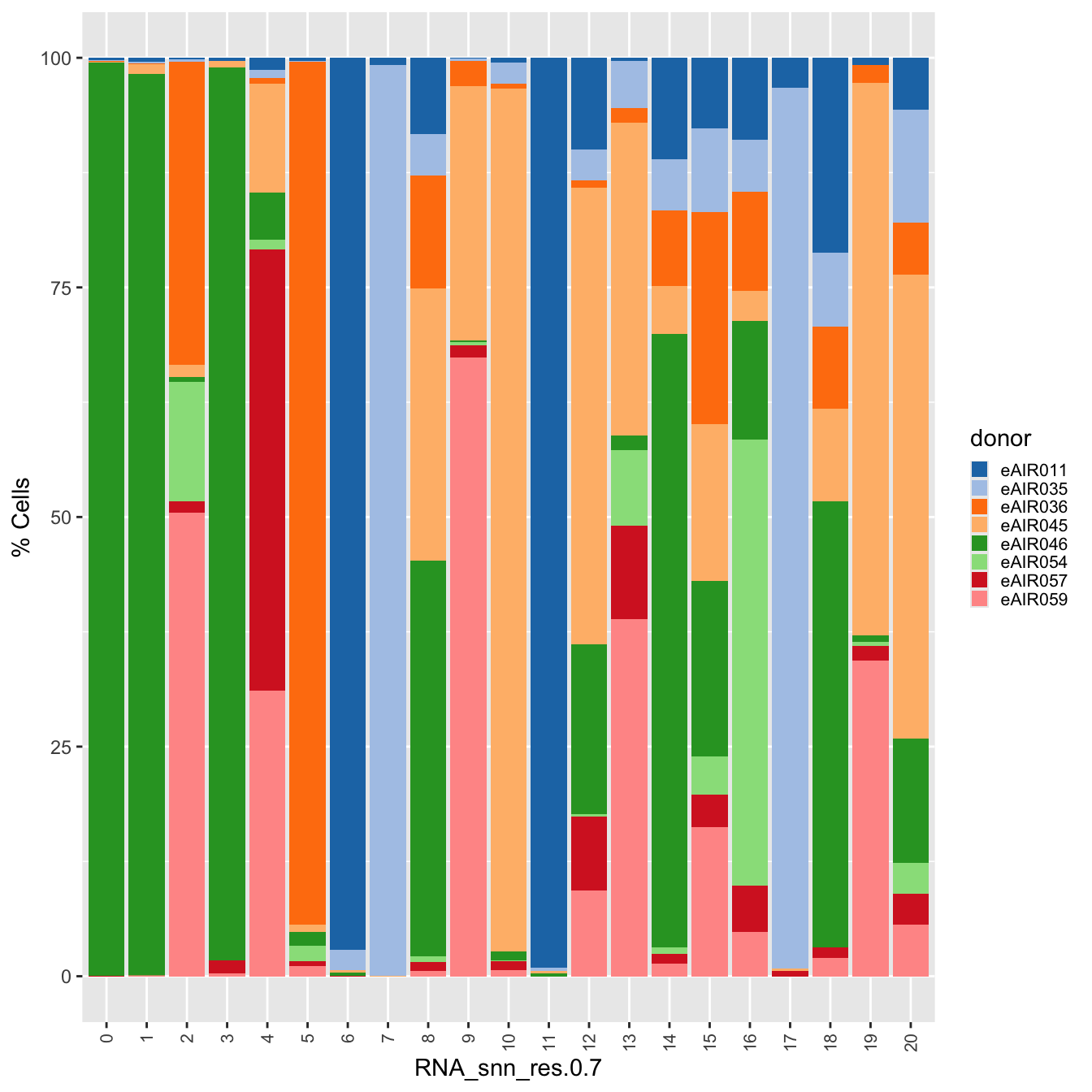
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
paed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8Calculating cluster 9Calculating cluster 10Calculating cluster 11Calculating cluster 12Calculating cluster 13Calculating cluster 14Calculating cluster 15Calculating cluster 16Calculating cluster 17Calculating cluster 18Calculating cluster 19Calculating cluster 20paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 10, wt = avg_log2FC) -> top10
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "THBS1" "APP" "SERPINB9" "SENP3" "ABCA1" "PRDX2"
[7] "DEFB1" "ENPP4" "VCAN" "CXCL2" "PLA1A" "MT-ND3"
[13] "RSRP1" "SDS" "E2F1" "SERPINB9" "SQSTM1" "NRP2"
[19] "MKI67" "CCL20" "PROK2" Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, ncol = 2, label = TRUE)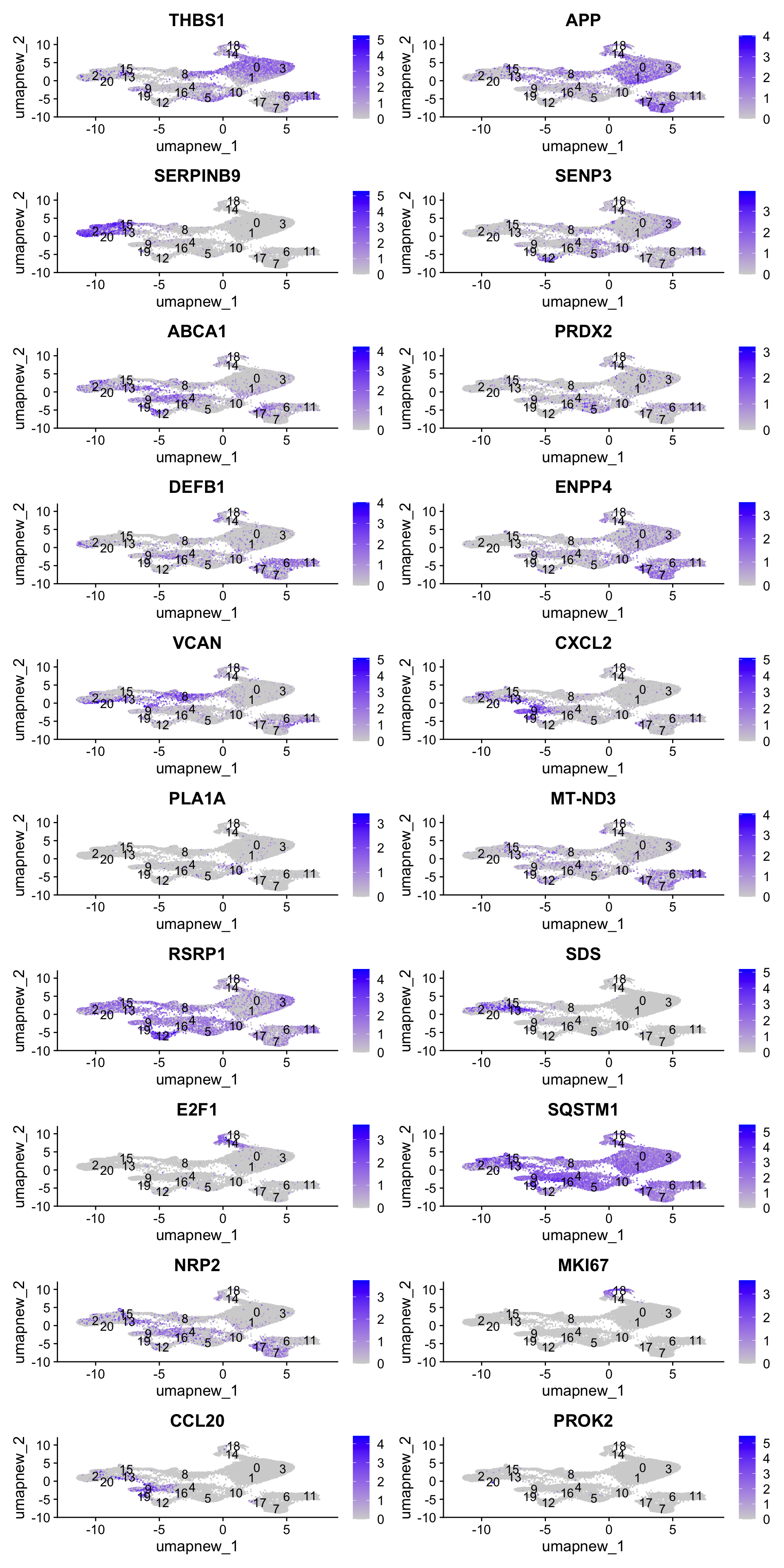
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.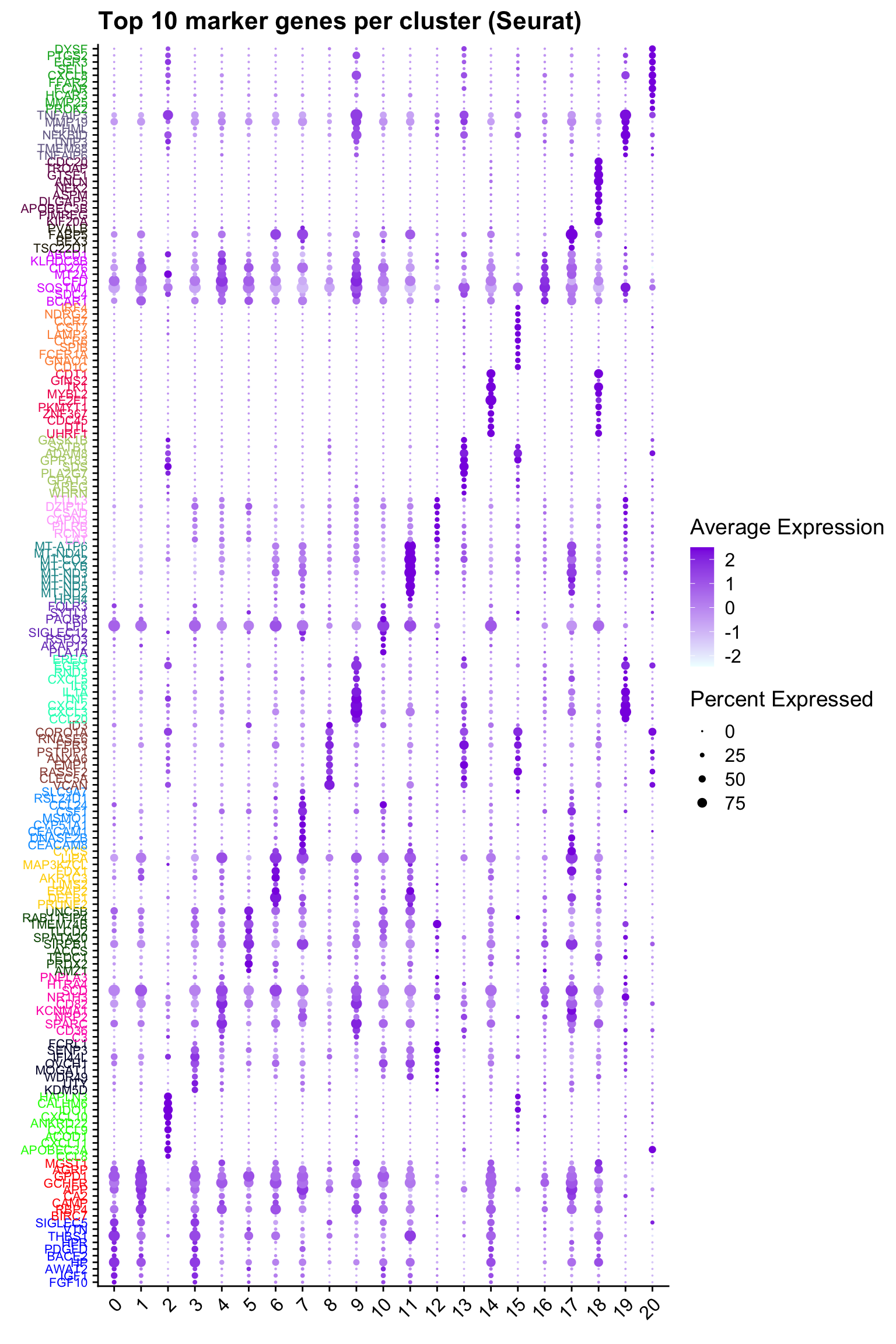
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_macro_population.withoutDecontX.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
}Save subclustered SEU object
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".macro_population.subclusters_without_DecontX.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Reclustering T/NK polulation
Here is the link to marker gene analysis of Macrophages in BAL (without ambient removal) BAL_Tcell_withoutDecontX_res.0.4
idx <- which(Idents(seu_obj) %in% "T/NK cells")
paed_sub <- seu_obj[,idx]
paed_subAn object of class Seurat
17529 features across 4631 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
2 dimensional reductions calculated: pca, umappaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9321
Number of communities: 4
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9048
Number of communities: 8
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8828
Number of communities: 9
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8660
Number of communities: 11
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8502
Number of communities: 11
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8359
Number of communities: 12
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8239
Number of communities: 13
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8136
Number of communities: 15
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8045
Number of communities: 17
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4631
Number of edges: 181992
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7955
Number of communities: 17
Elapsed time: 1 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)
clustree(paed_sub, prefix = "RNA_snn_res.")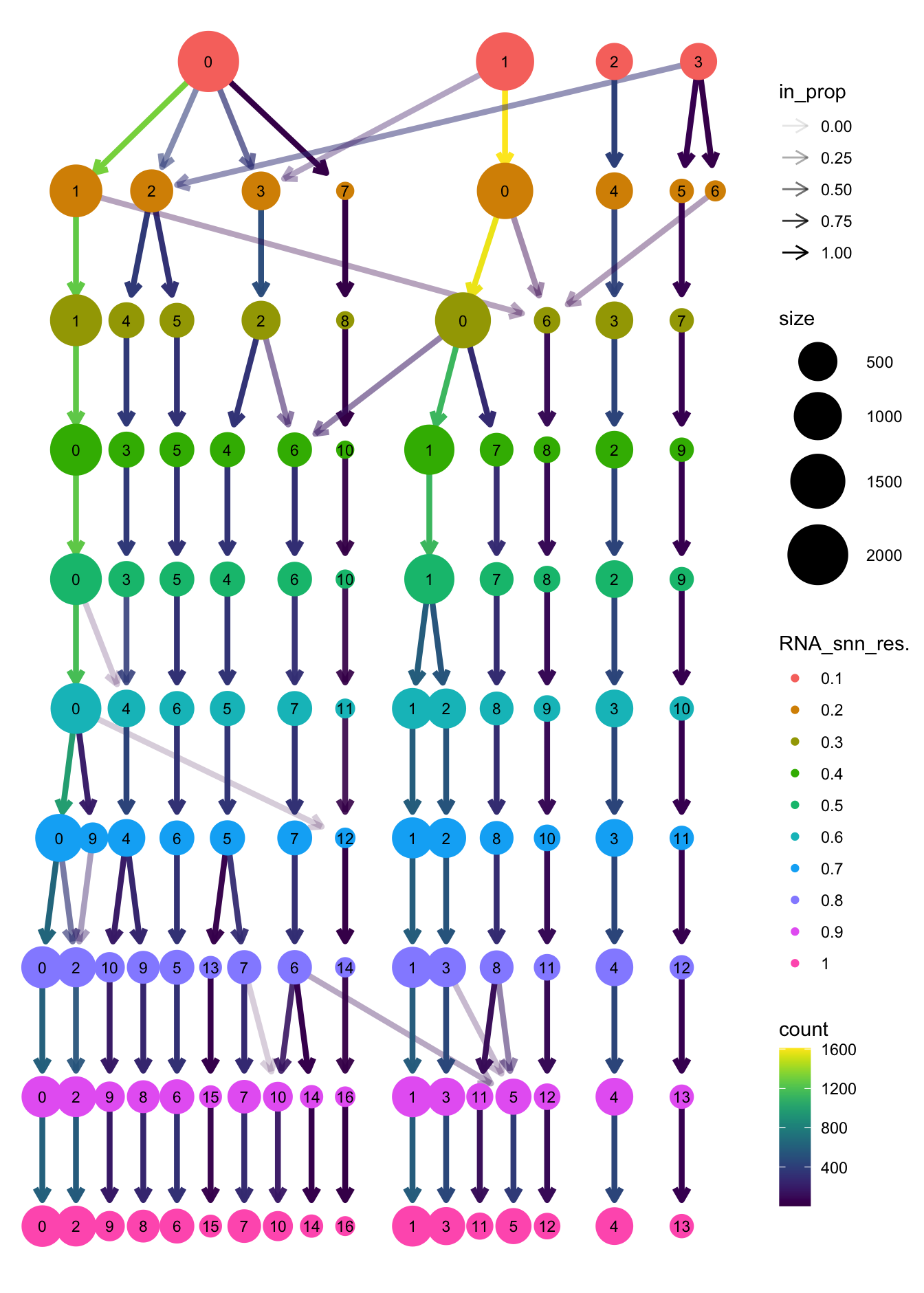
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(paed_sub$RNA_snn_res.0.4)
paed_sub$RNA_snn_res.0.4 <- factor(paed_sub$RNA_snn_res.0.4, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
paed_sub$cluster <- paed_sub$RNA_snn_res.0.4
Idents(paed_sub) <- paed_sub$clusterpaed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8Calculating cluster 9Calculating cluster 10paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "CCL5" "CD4" "NMUR1" "TCF7" "GZMB" "NCAM1" "MAF" "CXCR5"
[9] "CX3CR1" "KIT" "TFEC" Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, ncol = 2, label = TRUE)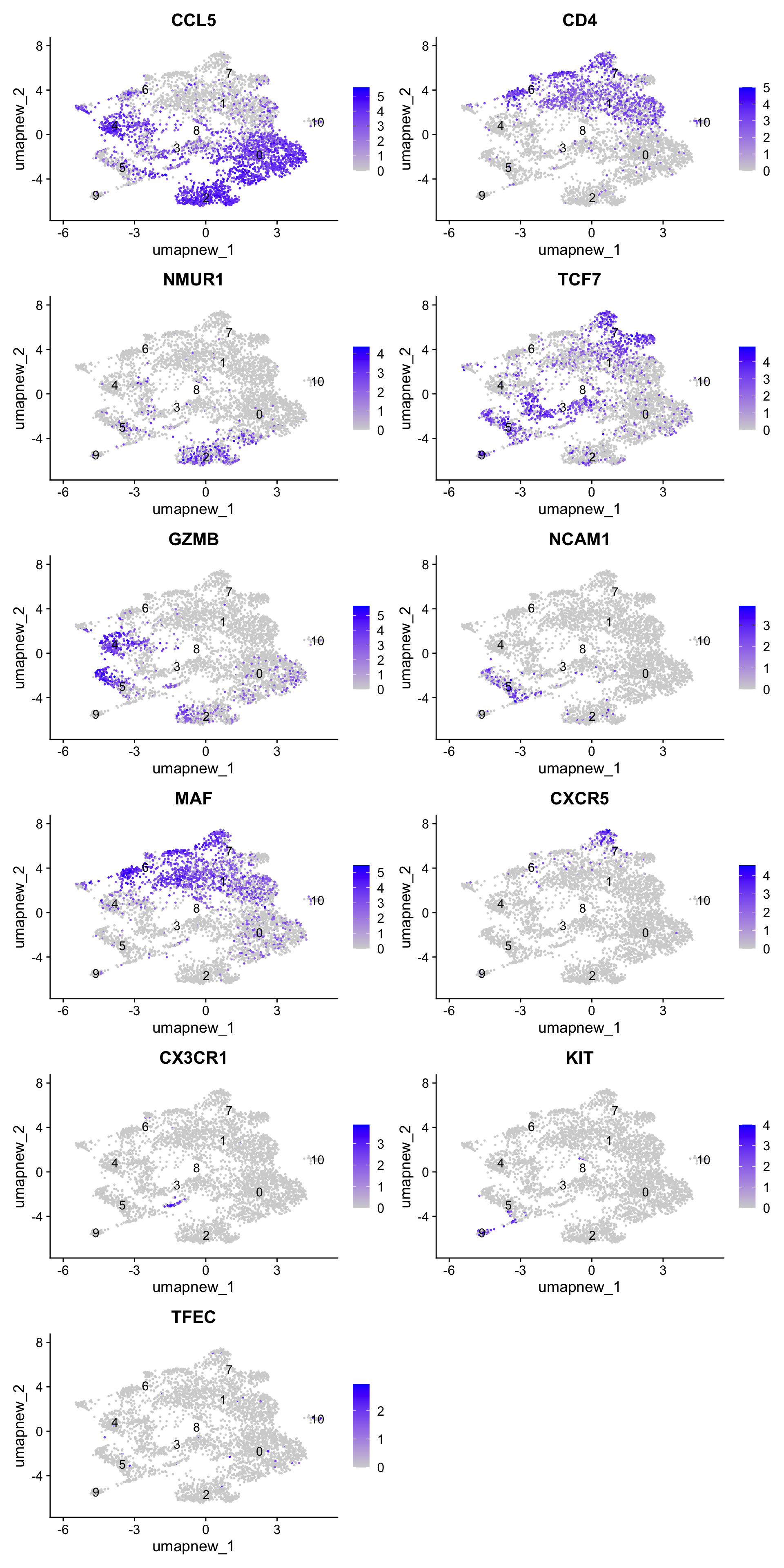
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.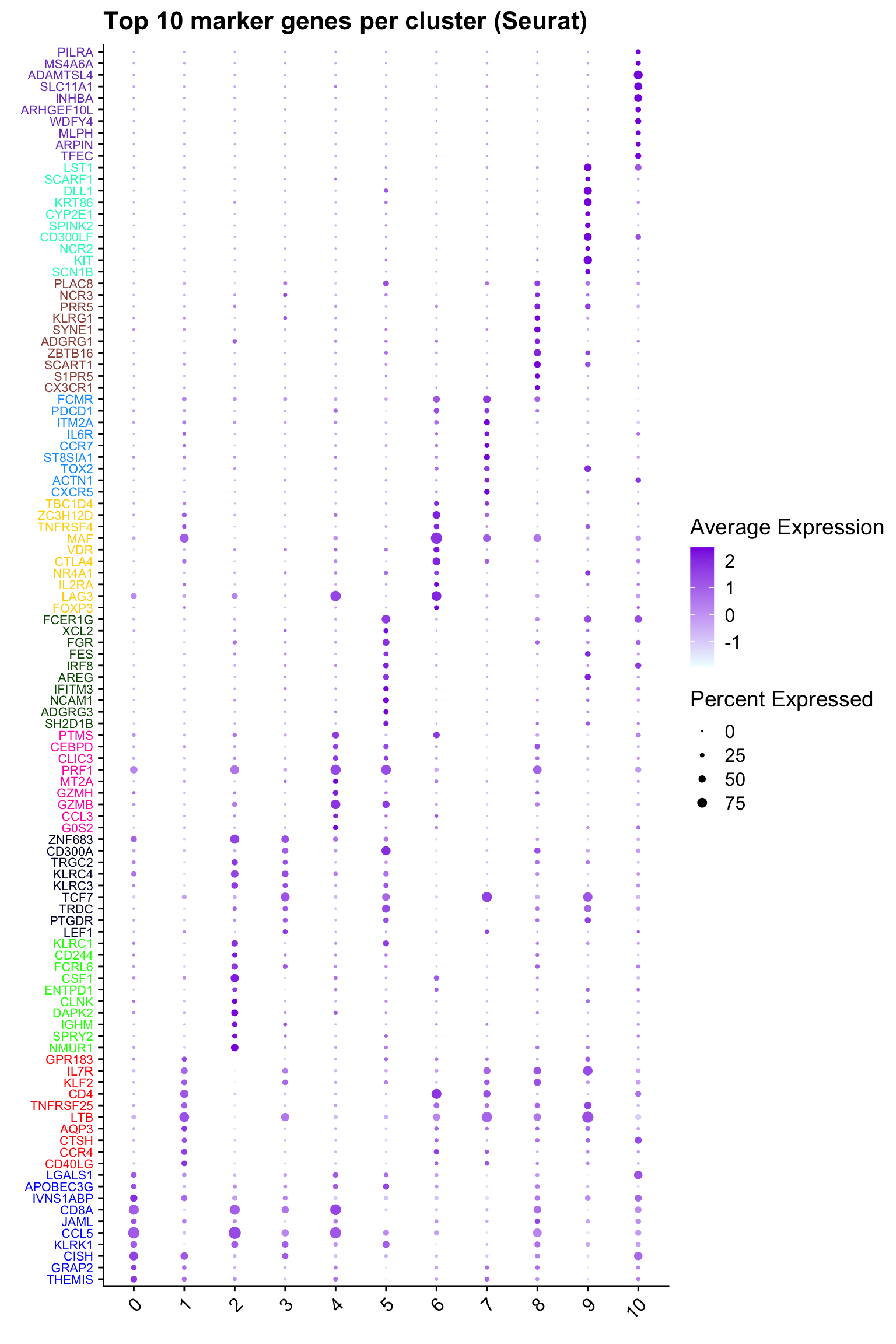
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_Tcell_population.withoutDecontX.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
}Save subclustered SEU object
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Tcell_population.subclusters_without_DecontX.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Reclustering Bcell polulation
Here is the link to marker gene analysis of Macrophages in BAL (without ambient removal) BAL_Bcell_withoutDecontX_res.0.4
idx <- which(Idents(seu_obj) %in% "B cells")
paed_sub <- seu_obj[,idx]
paed_subAn object of class Seurat
17529 features across 2176 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
2 dimensional reductions calculated: pca, umappaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9388
Number of communities: 4
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9042
Number of communities: 6
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8780
Number of communities: 7
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8571
Number of communities: 7
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8390
Number of communities: 7
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8231
Number of communities: 9
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8079
Number of communities: 9
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7956
Number of communities: 11
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7848
Number of communities: 12
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2176
Number of edges: 85004
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7742
Number of communities: 12
Elapsed time: 0 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)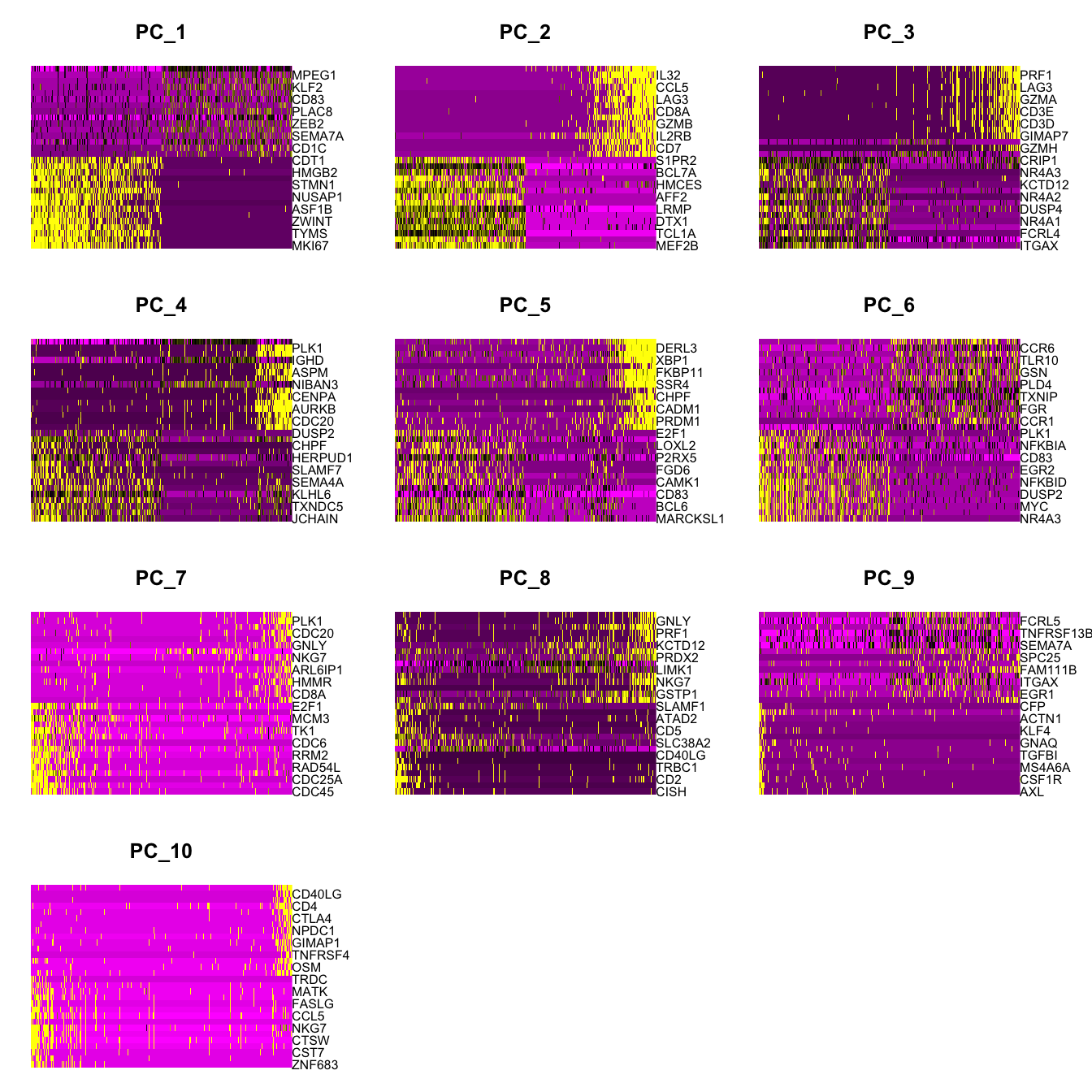
clustree(paed_sub, prefix = "RNA_snn_res.")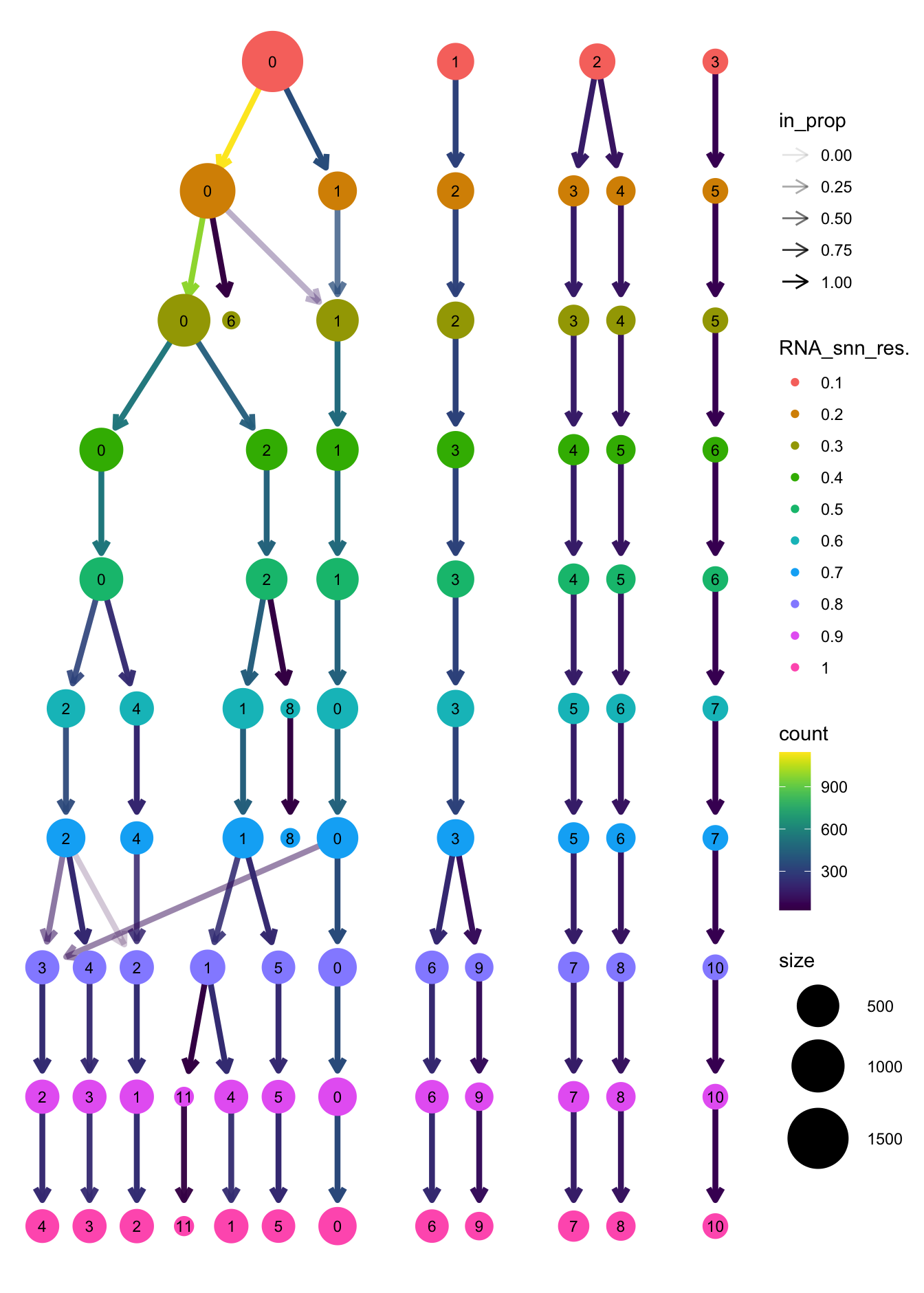
| Version | Author | Date |
|---|---|---|
| 0f07f72 | Gunjan Dixit | 2024-10-07 |
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(paed_sub$RNA_snn_res.0.4)
paed_sub$RNA_snn_res.0.4 <- factor(paed_sub$RNA_snn_res.0.4, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
paed_sub$cluster <- paed_sub$RNA_snn_res.0.4
Idents(paed_sub) <- paed_sub$clusterpaed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster[1] "ITGAX" "IGHD" "EGR3" "MEF2B" "KIFC1" "IL32" "HID1" Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, ncol = 2, label = TRUE)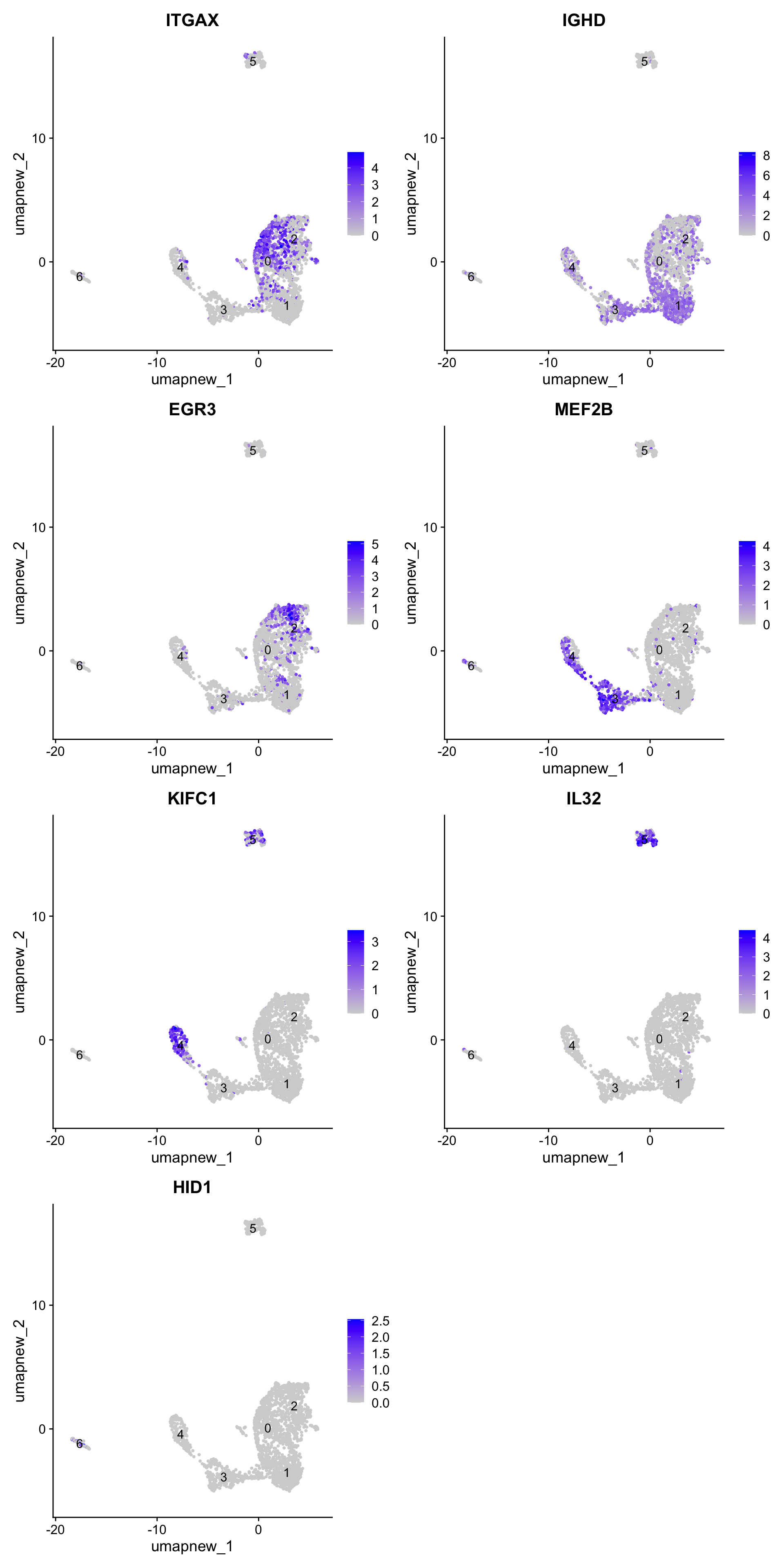
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.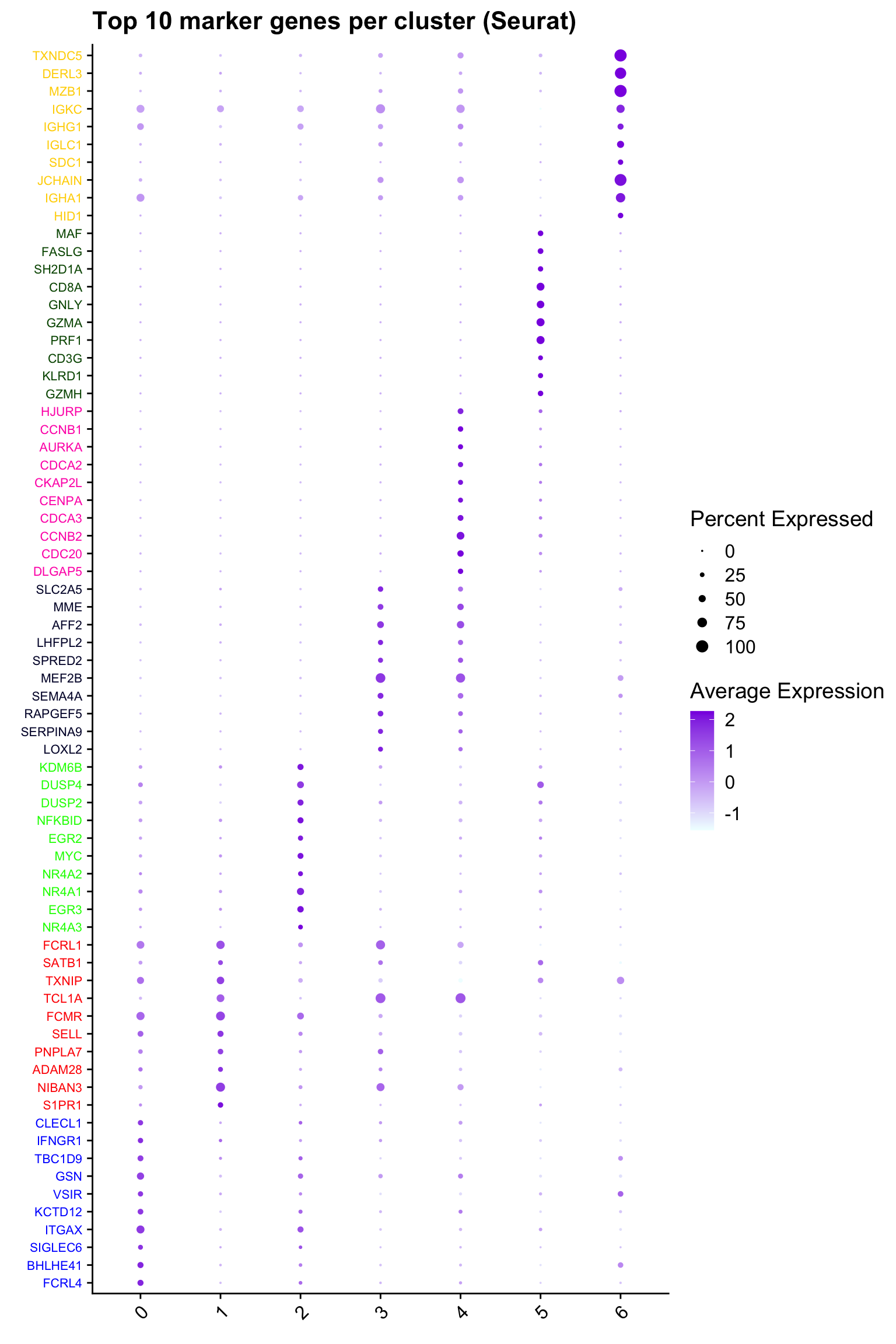
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_Bcell_population.withoutDecontX.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
}Save subclustered SEU object
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Bcell_population.subclusters_without_DecontX.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Compare two seurat objects
cl7_cells <- WhichCells(paed_sub, idents = 7)out3 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".macro_population.subclusters.SEU.rds"))
paed_sub1 <- readRDS(out3)DimPlot(paed_sub, reduction = "umap.new", group.by = "RNA_snn_res.0.7", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE , cells.highlight = cl7_cells)
DimPlot(paed_sub1, reduction = "umap.new", group.by = "RNA_snn_res.0.7", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE , cells.highlight = cl7_cells)
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.0.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] readxl_1.4.3 org.Hs.eg.db_3.18.0 AnnotationDbi_1.64.1
[4] IRanges_2.36.0 S4Vectors_0.40.2 Biobase_2.62.0
[7] BiocGenerics_0.48.1 speckle_1.2.0 edgeR_4.0.16
[10] limma_3.58.1 patchwork_1.2.0 data.table_1.15.0
[13] RColorBrewer_1.1-3 kableExtra_1.4.0 clustree_0.5.1
[16] ggraph_2.1.0 Seurat_5.0.1.9009 SeuratObject_5.0.1
[19] sp_2.1-3 glue_1.7.0 here_1.0.1
[22] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1
[25] dplyr_1.1.4 purrr_1.0.2 readr_2.1.5
[28] tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.0
[31] tidyverse_2.0.0 BiocStyle_2.30.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.3 matrixStats_1.2.0
[3] spatstat.sparse_3.0-3 bitops_1.0-7
[5] httr_1.4.7 tools_4.3.2
[7] sctransform_0.4.1 backports_1.4.1
[9] utf8_1.2.4 R6_2.5.1
[11] lazyeval_0.2.2 uwot_0.1.16
[13] withr_3.0.0 gridExtra_2.3
[15] progressr_0.14.0 cli_3.6.2
[17] spatstat.explore_3.2-6 fastDummies_1.7.3
[19] prismatic_1.1.1 labeling_0.4.3
[21] sass_0.4.8 spatstat.data_3.0-4
[23] ggridges_0.5.6 pbapply_1.7-2
[25] systemfonts_1.0.5 svglite_2.1.3
[27] parallelly_1.37.0 rstudioapi_0.15.0
[29] RSQLite_2.3.5 generics_0.1.3
[31] ica_1.0-3 spatstat.random_3.2-2
[33] Matrix_1.6-5 ggbeeswarm_0.7.2
[35] fansi_1.0.6 abind_1.4-5
[37] lifecycle_1.0.4 whisker_0.4.1
[39] yaml_2.3.8 SummarizedExperiment_1.32.0
[41] SparseArray_1.2.4 Rtsne_0.17
[43] paletteer_1.6.0 grid_4.3.2
[45] blob_1.2.4 promises_1.2.1
[47] crayon_1.5.2 miniUI_0.1.1.1
[49] lattice_0.22-5 cowplot_1.1.3
[51] KEGGREST_1.42.0 pillar_1.9.0
[53] knitr_1.45 GenomicRanges_1.54.1
[55] future.apply_1.11.1 codetools_0.2-19
[57] leiden_0.4.3.1 getPass_0.2-4
[59] vctrs_0.6.5 png_0.1-8
[61] spam_2.10-0 cellranger_1.1.0
[63] gtable_0.3.4 rematch2_2.1.2
[65] cachem_1.0.8 xfun_0.42
[67] S4Arrays_1.2.0 mime_0.12
[69] tidygraph_1.3.1 survival_3.5-8
[71] SingleCellExperiment_1.24.0 statmod_1.5.0
[73] ellipsis_0.3.2 fitdistrplus_1.1-11
[75] ROCR_1.0-11 nlme_3.1-164
[77] bit64_4.0.5 RcppAnnoy_0.0.22
[79] GenomeInfoDb_1.38.6 rprojroot_2.0.4
[81] bslib_0.6.1 irlba_2.3.5.1
[83] vipor_0.4.7 KernSmooth_2.23-22
[85] colorspace_2.1-0 DBI_1.2.2
[87] ggrastr_1.0.2 tidyselect_1.2.0
[89] processx_3.8.3 bit_4.0.5
[91] compiler_4.3.2 git2r_0.33.0
[93] xml2_1.3.6 DelayedArray_0.28.0
[95] plotly_4.10.4 checkmate_2.3.1
[97] scales_1.3.0 lmtest_0.9-40
[99] callr_3.7.5 digest_0.6.34
[101] goftest_1.2-3 spatstat.utils_3.0-4
[103] presto_1.0.0 rmarkdown_2.25
[105] XVector_0.42.0 htmltools_0.5.7
[107] pkgconfig_2.0.3 MatrixGenerics_1.14.0
[109] highr_0.10 fastmap_1.1.1
[111] rlang_1.1.3 htmlwidgets_1.6.4
[113] shiny_1.8.0 farver_2.1.1
[115] jquerylib_0.1.4 zoo_1.8-12
[117] jsonlite_1.8.8 RCurl_1.98-1.14
[119] magrittr_2.0.3 GenomeInfoDbData_1.2.11
[121] dotCall64_1.1-1 munsell_0.5.0
[123] Rcpp_1.0.12 viridis_0.6.5
[125] reticulate_1.35.0 stringi_1.8.3
[127] zlibbioc_1.48.0 MASS_7.3-60.0.1
[129] plyr_1.8.9 parallel_4.3.2
[131] listenv_0.9.1 ggrepel_0.9.5
[133] deldir_2.0-2 Biostrings_2.70.2
[135] graphlayouts_1.1.0 splines_4.3.2
[137] tensor_1.5 hms_1.1.3
[139] locfit_1.5-9.8 ps_1.7.6
[141] igraph_2.0.2 spatstat.geom_3.2-8
[143] RcppHNSW_0.6.0 reshape2_1.4.4
[145] evaluate_0.23 BiocManager_1.30.22
[147] tzdb_0.4.0 tweenr_2.0.3
[149] httpuv_1.6.14 RANN_2.6.1
[151] polyclip_1.10-6 future_1.33.1
[153] scattermore_1.2 ggforce_0.4.2
[155] xtable_1.8-4 RSpectra_0.16-1
[157] later_1.3.2 viridisLite_0.4.2
[159] beeswarm_0.4.0 memoise_2.0.1
[161] cluster_2.1.6 timechange_0.3.0
[163] globals_0.16.2