Differential Gene testing: Tonsils
IFN cells
George Howitt
February 07, 2025
Last updated: 2025-02-07
Checks: 6 1
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version e382550. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/RDS/
Ignored: output/.DS_Store
Ignored: output/CSV/.DS_Store
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Ignored: output/G000231_Neeland_batch9_1/
Ignored: output/RDS/
Ignored: output/plots/
Untracked files:
Untracked: analysis/03_Batch_Integration.Rmd
Untracked: analysis/Age_proportions.Rmd
Untracked: analysis/Age_proportions_AllBatches.Rmd
Untracked: analysis/All_Batches_QCExploratory_v2.Rmd
Untracked: analysis/All_metadata.Rmd
Untracked: analysis/Annotation_BAL.Rmd
Untracked: analysis/Annotation_Bronchial_brushings.Rmd
Untracked: analysis/Annotation_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Adenoids.Rmd
Untracked: analysis/BatchCorrection_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Tonsils.Rmd
Untracked: analysis/Batch_Integration_&_Downstream_analysis.Rmd
Untracked: analysis/Batch_correction_&_Downstream.Rmd
Untracked: analysis/Cell_cycle_regression.Rmd
Untracked: analysis/Clustering_Tonsils_v2.Rmd
Untracked: analysis/DGE_TonsilAtlas.Rmd
Untracked: analysis/DGE_Tonsils_Naive_B.Rmd
Untracked: analysis/DGE_analysis_George.Rmd
Untracked: analysis/Master_metadata.Rmd
Untracked: analysis/Pediatric_Vs_Adult_Atlases.Rmd
Untracked: analysis/Preprocessing_Batch1_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch2_Tonsils.Rmd
Untracked: analysis/Preprocessing_Batch3_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch4_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch5_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch6_BAL.Rmd
Untracked: analysis/Preprocessing_Batch7_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch8_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch9_Tonsils.Rmd
Untracked: analysis/TonsilsVsAdenoids.Rmd
Untracked: analysis/cell_cycle_regression.R
Untracked: analysis/testing_age_all.Rmd
Untracked: code/pseudobulk_analysis.R
Untracked: data/Cell_labels_Gunjan_v2/
Untracked: data/Cell_labels_Mel/
Untracked: data/Cell_labels_Mel_v2/
Untracked: data/Cell_labels_Mel_v3/
Untracked: data/Cell_labels_modified_Gunjan/
Untracked: data/Gene_sets/
Untracked: data/Hs.c2.cp.reactome.v7.1.entrez.rds
Untracked: data/Raw_feature_bc_matrix/
Untracked: data/cell_labels_Mel_v4_Dec2024/
Untracked: data/celltypes_Mel_GD_v3.xlsx
Untracked: data/celltypes_Mel_GD_v4_no_dups.xlsx
Untracked: data/celltypes_Mel_modified.xlsx
Untracked: data/celltypes_Mel_v2.csv
Untracked: data/celltypes_Mel_v2.xlsx
Untracked: data/celltypes_Mel_v2_MN.xlsx
Untracked: data/celltypes_for_mel_MN.xlsx
Untracked: data/col_palette.xlsx
Untracked: data/earlyAIR_sample_sheets_combined.xlsx
Untracked: figure/DGE_Tonsils_CD8.Rmd/
Untracked: output/CSV/All_tissues.propeller.xlsx
Untracked: output/CSV/Bronchial_brushings/
Untracked: output/CSV/Bronchial_brushings_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/
Untracked: output/CSV/G000231_Neeland_Adenoids.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Bronchial_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Nasal_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Tonsils.propeller.xlsx
Untracked: output/CSV/Nasal_brushings/
Untracked: output/DGE/Adenoids_B_memory/
Untracked: output/DGE/Adenoids_CD4/
Untracked: output/DGE/Adenoids_CD8/
Untracked: output/DGE/Adenoids_Cycling_GCB/
Untracked: output/DGE/Adenoids_IFN/
Untracked: output/DGE/Tonsil_Atlas_CD4/
Untracked: output/DGE/Tonsils/DGE_Age_summary_Tonsils.xlsx
Untracked: output/DGE/Tonsils/~$DGE_Age_summary_Tonsils.xlsx
Untracked: output/DGE/Tonsils_CD4/
Untracked: output/DGE/Tonsils_CD8/
Untracked: output/DGE/Tonsils_IFN/
Untracked: test_col.csv
Untracked: test_col.txt
Untracked: test_col.xlsx
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Modified: analysis/00_AllBatches_overview.Rmd
Modified: analysis/01_QC_emptyDrops.Rmd
Modified: analysis/02_QC_exploratoryPlots.Rmd
Modified: analysis/Adenoids.Rmd
Modified: analysis/Adenoids_v2.Rmd
Modified: analysis/Age_modeling.Rmd
Modified: analysis/AllBatches_QCExploratory.Rmd
Modified: analysis/BAL.Rmd
Modified: analysis/BAL_v2.Rmd
Modified: analysis/Bronchial_brushings.Rmd
Modified: analysis/Bronchial_brushings_v2.Rmd
Modified: analysis/DGE_Tonsils_CD4.Rmd
Deleted: analysis/DGE_Tonsils_Cycling_GCB.Rmd
Modified: analysis/DGE_Tonsils_IFN.Rmd
Modified: analysis/DGE_Tonsils_Memory_B.Rmd
Modified: analysis/Nasal_brushings.Rmd
Modified: analysis/Nasal_brushings_v2.Rmd
Modified: analysis/Subclustering_Adenoids.Rmd
Modified: analysis/Subclustering_BAL.Rmd
Modified: analysis/Subclustering_Bronchial_brushings.Rmd
Modified: analysis/Subclustering_Nasal_brushings.Rmd
Modified: analysis/Subclustering_Tonsils.Rmd
Modified: analysis/Tonsils.Rmd
Modified: analysis/Tonsils_v2.Rmd
Modified: analysis/index.Rmd
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c9.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c9.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/DGE_Tonsils_IFN.Rmd) and
HTML (docs/DGE_Tonsils_IFN.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 600b673 | Gunjan Dixit | 2025-02-05 | Added DGE analysis for Tonsils |
| html | 600b673 | Gunjan Dixit | 2025-02-05 | Added DGE analysis for Tonsils |
suppressPackageStartupMessages({
library(here)
library(glue)
library(patchwork)
library(Seurat)
library(dplyr)
library(tidyverse)
library(gridExtra)
library(paletteer)
library(viridis)
library(tidyverse)
library(scran)
library(scater)
library(ggridges)
library(speckle)
library(edgeR)
library(limma)
library(knitr)
library(BiocStyle)
library(org.Hs.eg.db)
library(Glimma)
})Load Data
data_path <- here("output/RDS/AllBatches_Annotation_SEUs_v2/")
tissue <- "Tonsils"
seu <- readRDS(paste0(data_path, "G000231_Neeland_", tissue, ".annotated_clusters.SEU.rds"))seu <- JoinLayers(seu)Convert from Seurat to SingleCellExperiment object
sce <- SingleCellExperiment(list(counts = seu@assays$RNA@layers$counts),
colData = seu@meta.data)
rownames(sce) <- rownames(seu)What cell populations are present?
table(sce$cell_labels_v2)
CD4 effector CD4 TCM
2944 3115
CD4 TFH CD4 TN
13486 10211
CD4 Treg CD4 Treg-eff
2035 3248
CD8 TF CD8 TN
4666 4221
Cycling GCB Cycling T
4243 536
Double negative T DZ early Sphase
1268 5716
DZ G2Mphase DZ GCB
9872 7913
DZ late Sphase DZtoLZ GCB transition
3909 8341
Early GC-committed NBC Early MBC
5260 8234
Early PC precursor Epithelial cells
1354 392
Follicular dendritic cells Gamma delta T
1688 1657
GC-commited metabolic activation GCB-IFN
1693 324
MAIT cells Mast cells
511 66
Memory B cells Monocytes/macrophages
21730 3004
Naïve B cell-IFN Naïve B cells
7343 38686
Naïve B cells activated Neutrophils
6632 552
NK cells Plasma B cells
876 9121
Plasmacytoid DCs Pre-BCRi II
429 2743
Pre-T cells T-IFN
93 1917
TFH-LZ-GC
8048 Subset the cells of interest and pseudobulk
Pseudobulk
celltype = "IFN"
celltypes_to_subset <- c("Naïve B cell-IFN","GCB-IFN ", "T-IFN")
pb <- aggregateAcrossCells(sce[, sce$cell_labels_v2 %in% celltypes_to_subset],
id = colData(sce[, sce$cell_labels_v2 %in% celltypes_to_subset])[, c("sample_id")])
pbclass: SingleCellExperiment
dim: 17566 32
metadata(0):
assays(1): counts
rownames(17566): SAMD11 NOC2L ... HSFX4 DAZ2
rowData names(0):
colnames(32): s017 s018 ... s152 s153
colData names(45): donor_id sample_id ... ids ncells
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):Remove samples with fewer than 50 cells
pb <- pb[, pb$ncells>=50]
pbclass: SingleCellExperiment
dim: 17566 32
metadata(0):
assays(1): counts
rownames(17566): SAMD11 NOC2L ... HSFX4 DAZ2
rowData names(0):
colnames(32): s017 s018 ... s152 s153
colData names(45): donor_id sample_id ... ids ncells
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):Clean up metadata
pb <- SingleCellExperiment(list(counts = counts(pb)),
colData = colData(pb) %>%
data.frame %>%
dplyr::select(c("sample_id","donor_id", "age_years", "sex", "sample_barcode", "tissue", "batch_name", "ncells")) %>%
DataFrame) %>%
addPerCellQCMetricsAdd categorical age variable
pb$age_category <- ifelse(pb$age_years <= 5, 'preschool',
ifelse(pb$age_years > 5 & pb$age_years <= 11, 'early_childhood',
ifelse(pb$age_years > 11, 'adolescent', NA)))ggplot(colData(pb) %>% data.frame) +
geom_violin(aes(x = sex, y = age_years, fill = sex),
position = 'dodge') +
geom_boxplot(aes(x = sex, y = age_years), width = 0.25) +
ggtitle(tissue)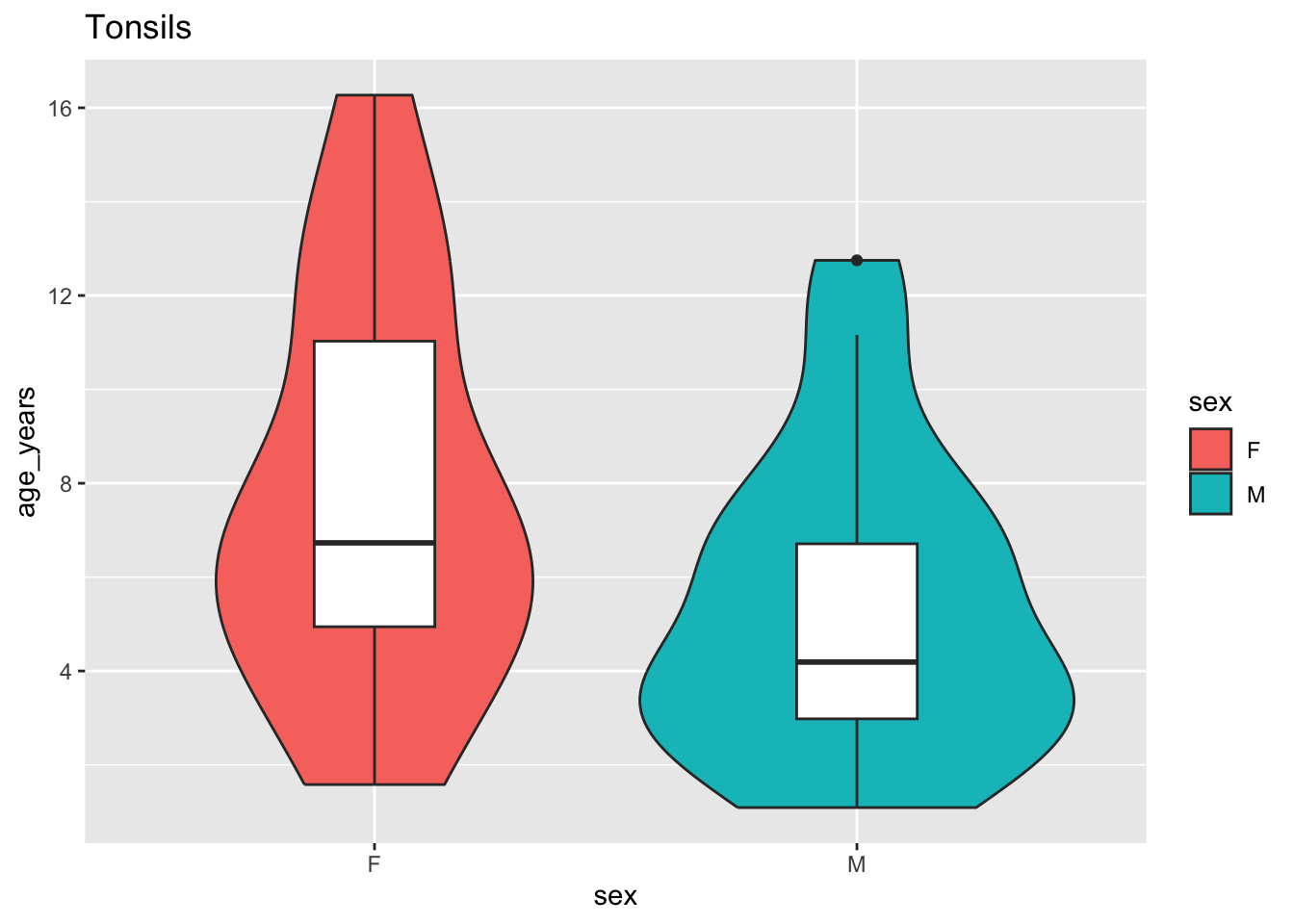
| Version | Author | Date |
|---|---|---|
| 600b673 | Gunjan Dixit | 2025-02-05 |
Perform lognorm and PCA
pb <- logNormCounts(pb) %>%
runPCA()Warning in check_numbers(k = k, nu = nu, nv = nv, limit = min(dim(x)) - : more
singular values/vectors requested than availableWarning in (function (A, nv = 5, nu = nv, maxit = 1000, work = nv + 7, reorth =
TRUE, : You're computing too large a percentage of total singular values, use a
standard svd instead.limma-voom analysis
Make a DGEList object
dge <- DGEList(counts = counts(pb),
samples = colData(pb) %>% data.frame)Filter unexpressed genes
keep <- rowSums(dge$counts) > 0
dge <- dge[keep, ]
dim(dge)[1] 14530 32dge <- calcNormFactors(dge)MDS plot
Using Glimma to look at MDS plots
glimmaMDS(dge)Make design matrix
design <- model.matrix(~dge$samples$age_years + dge$samples$sex + dge$samples$batch_name)Run limma-voom
v <- voom(dge, design, plot = T)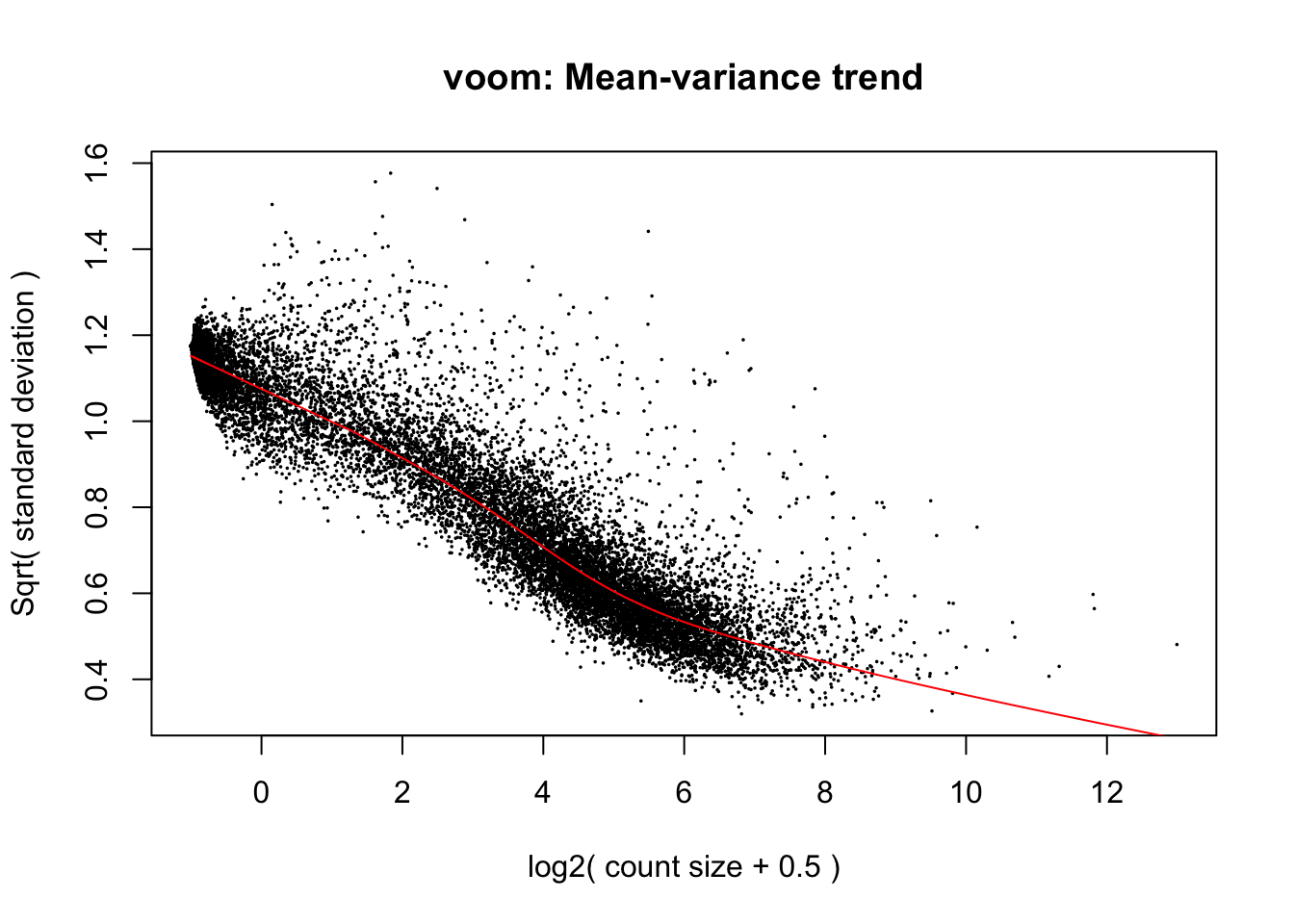
| Version | Author | Date |
|---|---|---|
| 600b673 | Gunjan Dixit | 2025-02-05 |
fit <- lmFit(v, design)
fit <- eBayes(fit)dt <- decideTests(fit)
summary(dt) (Intercept) dge$samples$age_years dge$samples$sexM
Down 6 1 6
NotSig 4080 14527 14518
Up 10444 2 6
dge$samples$batch_nameG000231_batch9
Down 1095
NotSig 12384
Up 1051Interactive volcano plot
coef = "dge$samples$age_years"
glimmaVolcano(fit, dge = dge, coef = coef, groups = dge$samples$age_category)Warning in buildXYData(table, status, main, display.columns, anno, counts, :
count transform requested but not all count values are integers.camera gene-set testing
Make gene-set lists from files
convert_gmt_to_list <- function(file_path){
# Read the file content
lines <- readLines(file_path)
# Pre-allocate the list with the number of lines in the file
gene_sets <- vector("list", length(lines))
# Loop over each line to process it
for (i in seq_along(lines)) {
# Split the line by tabs
elements <- strsplit(lines[i], "\t")[[1]]
# The first element is the name of the gene set
gene_set_name <- elements[1]
# The rest are the Entrez IDs (after the URL)
entrez_ids <- elements[-(1:2)]
# Store the gene set name and the vector of Entrez IDs
names(gene_sets)[i] <- gene_set_name
gene_sets[[i]] <- entrez_ids
}
gene_sets
}Hs.c2.all <- convert_gmt_to_list(here("data/Gene_sets/c2.all.v2024.1.Hs.entrez.gmt"))
Hs.h.all <- convert_gmt_to_list(here("data/Gene_sets/h.all.v2024.1.Hs.entrez.gmt"))
Hs.c5.all <- convert_gmt_to_list(here("data/Gene_sets/c5.all.v2024.1.Hs.entrez.gmt"))
gene_sets_list <- list(HALLMARK = Hs.h.all,
GO = Hs.c5.all,
REACTOME = Hs.c2.all[str_detect(names(Hs.c2.all), "REACTOME")],
WP = Hs.c2.all[str_detect(names(Hs.c2.all), "^WP")])library(org.Hs.eg.db)
gns <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = rownames(fit),
column = c("ENTREZID"),
keytype = "SYMBOL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnsgene_set_test_camera <- function(gene_sets_list, gns, lrt, statistic, cellDir){
cam_list <- lapply(seq_along(gene_sets_list), function(i){
id <- ids2indices(gene_sets_list[[i]], unname(gns[rownames(lrt)]))
tmp <- cameraPR(statistic, id)
write.table(tmp %>%
data.frame %>%
rownames_to_column(var = "Set"),
file = file.path(cellDir, glue("CAM.{names(gene_sets_list[i])}.csv")),
sep = ",", quote = F, col.names = NA)
tmp
})
names(cam_list) <- names(gene_sets_list)
cam_list
}output_path <- here("output/DGE", tissue)
output_dir <- file.path(output_path, glue("Tonsils_{celltype}"))
dir.create(output_dir, recursive = TRUE, showWarnings = FALSE)
test_set <- gene_set_test_camera(gene_sets_list, gns, fit, fit$t[,2], cellDir = output_dir)top_camera_sets <- function(results_list, num = 10){
lapply(seq_along(results_list), function(i){
results_list[[i]] %>%
data.frame %>%
dplyr::slice(1:min(num, n())) %>%
rownames_to_column(var = "Set") %>%
mutate(Type = names(results_list)[i],
Rank = 1:min(num, n()))
}) %>%
bind_rows %>%
mutate(Set = str_wrap(str_replace_all(Set, "_", " "), width = 75),
Set = str_remove_all(Set, "GO |REACTOME |HALLMARK |WP ")) %>%
ggplot(aes(x = -log10(FDR), y = fct_reorder(Set, -Rank),
colour = Direction)) +
geom_point(aes(size = NGenes)) +
facet_wrap(~Type, ncol = 1, scales = "free_y") +
geom_vline(xintercept = -log10(0.05),
linetype = "dashed") +
#scale_colour_manual(values = pal) +
labs(y = "Gene set") +
theme_classic(base_size = 10) +
ggtitle("Camera gene set analysis")
}top_camera_sets(test_set, num = 10)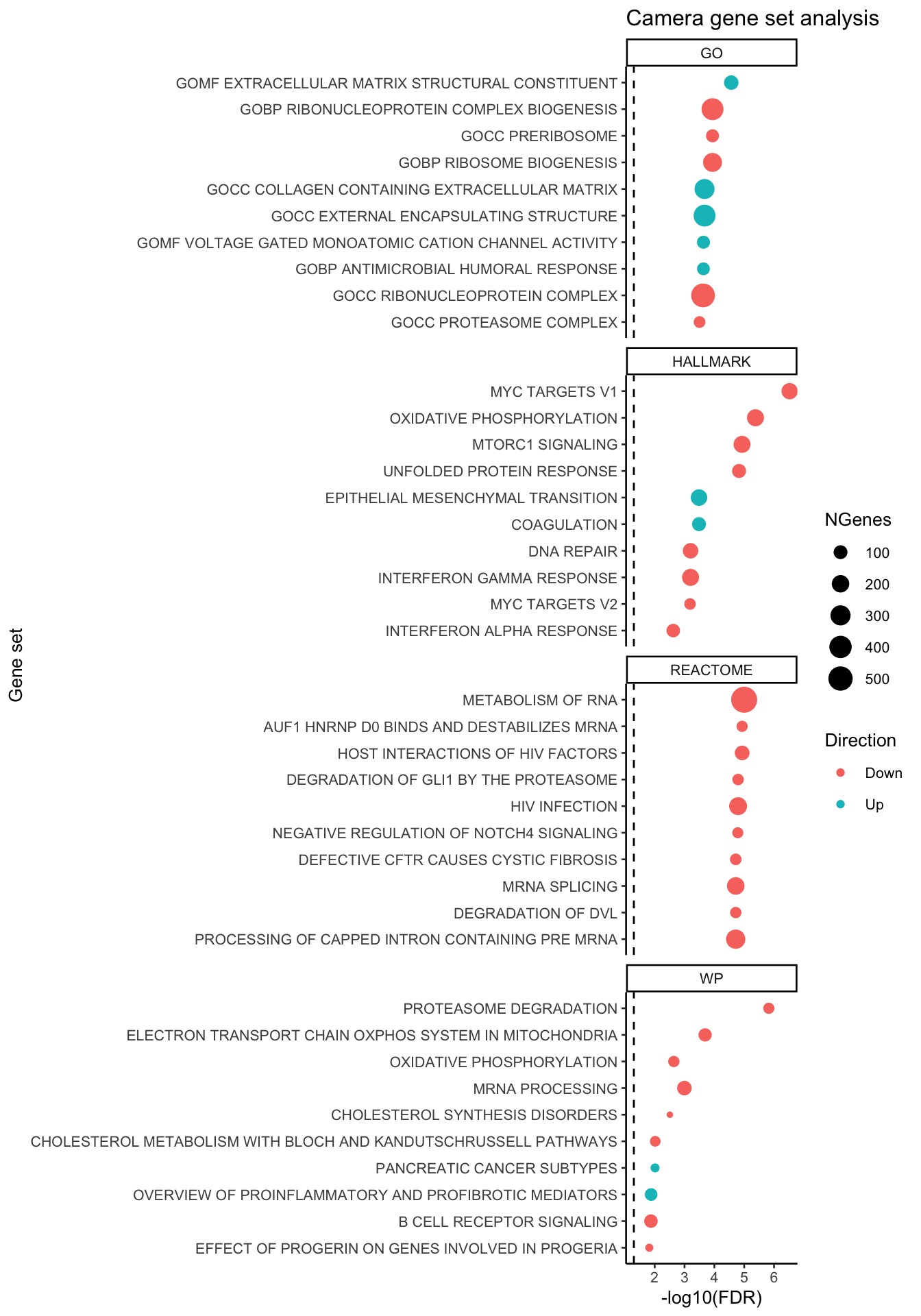
| Version | Author | Date |
|---|---|---|
| 600b673 | Gunjan Dixit | 2025-02-05 |
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.3
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] Glimma_2.12.0 org.Hs.eg.db_3.18.0
[3] AnnotationDbi_1.64.1 BiocStyle_2.30.0
[5] knitr_1.45 edgeR_4.0.16
[7] limma_3.58.1 speckle_1.2.0
[9] ggridges_0.5.6 scater_1.30.1
[11] scran_1.30.2 scuttle_1.12.0
[13] SingleCellExperiment_1.24.0 SummarizedExperiment_1.32.0
[15] Biobase_2.62.0 GenomicRanges_1.54.1
[17] GenomeInfoDb_1.38.6 IRanges_2.36.0
[19] S4Vectors_0.40.2 BiocGenerics_0.48.1
[21] MatrixGenerics_1.14.0 matrixStats_1.2.0
[23] viridis_0.6.5 viridisLite_0.4.2
[25] paletteer_1.6.0 gridExtra_2.3
[27] lubridate_1.9.3 forcats_1.0.0
[29] stringr_1.5.1 purrr_1.0.2
[31] readr_2.1.5 tidyr_1.3.1
[33] tibble_3.2.1 ggplot2_3.5.0
[35] tidyverse_2.0.0 dplyr_1.1.4
[37] Seurat_5.0.1.9009 SeuratObject_5.0.1
[39] sp_2.1-3 patchwork_1.2.0
[41] glue_1.7.0 here_1.0.1
[43] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.3 spatstat.sparse_3.0-3
[3] bitops_1.0-7 httr_1.4.7
[5] RColorBrewer_1.1-3 tools_4.3.2
[7] sctransform_0.4.1 utf8_1.2.4
[9] R6_2.5.1 lazyeval_0.2.2
[11] uwot_0.1.16 withr_3.0.0
[13] progressr_0.14.0 cli_3.6.2
[15] spatstat.explore_3.2-6 fastDummies_1.7.3
[17] labeling_0.4.3 sass_0.4.8
[19] spatstat.data_3.0-4 pbapply_1.7-2
[21] parallelly_1.37.0 rstudioapi_0.15.0
[23] RSQLite_2.3.5 generics_0.1.3
[25] ica_1.0-3 spatstat.random_3.2-2
[27] Matrix_1.6-5 ggbeeswarm_0.7.2
[29] fansi_1.0.6 abind_1.4-5
[31] lifecycle_1.0.4 whisker_0.4.1
[33] yaml_2.3.8 SparseArray_1.2.4
[35] Rtsne_0.17 grid_4.3.2
[37] blob_1.2.4 promises_1.2.1
[39] dqrng_0.3.2 crayon_1.5.2
[41] miniUI_0.1.1.1 lattice_0.22-5
[43] beachmat_2.18.1 cowplot_1.1.3
[45] KEGGREST_1.42.0 pillar_1.9.0
[47] metapod_1.10.1 future.apply_1.11.1
[49] codetools_0.2-19 leiden_0.4.3.1
[51] getPass_0.2-4 data.table_1.15.0
[53] vctrs_0.6.5 png_0.1-8
[55] spam_2.10-0 gtable_0.3.4
[57] rematch2_2.1.2 cachem_1.0.8
[59] xfun_0.42 S4Arrays_1.2.0
[61] mime_0.12 survival_3.5-8
[63] statmod_1.5.0 bluster_1.12.0
[65] ellipsis_0.3.2 fitdistrplus_1.1-11
[67] ROCR_1.0-11 nlme_3.1-164
[69] bit64_4.0.5 RcppAnnoy_0.0.22
[71] rprojroot_2.0.4 bslib_0.6.1
[73] irlba_2.3.5.1 vipor_0.4.7
[75] KernSmooth_2.23-22 colorspace_2.1-0
[77] DBI_1.2.2 DESeq2_1.42.1
[79] tidyselect_1.2.0 processx_3.8.3
[81] bit_4.0.5 compiler_4.3.2
[83] git2r_0.33.0 BiocNeighbors_1.20.2
[85] DelayedArray_0.28.0 plotly_4.10.4
[87] scales_1.3.0 lmtest_0.9-40
[89] callr_3.7.5 digest_0.6.34
[91] goftest_1.2-3 spatstat.utils_3.0-4
[93] rmarkdown_2.25 XVector_0.42.0
[95] htmltools_0.5.7 pkgconfig_2.0.3
[97] sparseMatrixStats_1.14.0 highr_0.10
[99] fastmap_1.1.1 rlang_1.1.3
[101] htmlwidgets_1.6.4 shiny_1.8.0
[103] DelayedMatrixStats_1.24.0 farver_2.1.1
[105] jquerylib_0.1.4 zoo_1.8-12
[107] jsonlite_1.8.8 BiocParallel_1.36.0
[109] BiocSingular_1.18.0 RCurl_1.98-1.14
[111] magrittr_2.0.3 GenomeInfoDbData_1.2.11
[113] dotCall64_1.1-1 munsell_0.5.0
[115] Rcpp_1.0.12 reticulate_1.35.0
[117] stringi_1.8.3 zlibbioc_1.48.0
[119] MASS_7.3-60.0.1 plyr_1.8.9
[121] parallel_4.3.2 listenv_0.9.1
[123] ggrepel_0.9.5 deldir_2.0-2
[125] Biostrings_2.70.2 splines_4.3.2
[127] tensor_1.5 hms_1.1.3
[129] locfit_1.5-9.8 ps_1.7.6
[131] igraph_2.0.2 spatstat.geom_3.2-8
[133] RcppHNSW_0.6.0 reshape2_1.4.4
[135] ScaledMatrix_1.10.0 evaluate_0.23
[137] BiocManager_1.30.22 tzdb_0.4.0
[139] httpuv_1.6.14 RANN_2.6.1
[141] polyclip_1.10-6 future_1.33.1
[143] scattermore_1.2 rsvd_1.0.5
[145] xtable_1.8-4 RSpectra_0.16-1
[147] later_1.3.2 memoise_2.0.1
[149] beeswarm_0.4.0 cluster_2.1.6
[151] timechange_0.3.0 globals_0.16.2