DGE_earlyAIR_tAtlas_CD4_TCM
2025-02-26
Last updated: 2025-02-28
Checks: 6 1
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version db355f3. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/RDS/
Ignored: output/.DS_Store
Ignored: output/CAM_results/
Ignored: output/CSV/.DS_Store
Ignored: output/DGE/.DS_Store
Ignored: output/DGE/Tonsils/RUV_Tonsils_Double negative T/
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Ignored: output/G000231_Neeland_batch9_1/
Ignored: output/RDS/
Ignored: output/plots/
Untracked files:
Untracked: VennDiagram.2025-02-21_13-16-58.873484.log
Untracked: analysis/03_Batch_Integration.Rmd
Untracked: analysis/Age_proportions.Rmd
Untracked: analysis/Age_proportions_AllBatches.Rmd
Untracked: analysis/All_Batches_QCExploratory_v2.Rmd
Untracked: analysis/All_metadata.Rmd
Untracked: analysis/Annotation_BAL.Rmd
Untracked: analysis/Annotation_Bronchial_brushings.Rmd
Untracked: analysis/Annotation_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Adenoids.Rmd
Untracked: analysis/BatchCorrection_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Tonsils.Rmd
Untracked: analysis/Batch_Integration_&_Downstream_analysis.Rmd
Untracked: analysis/Batch_correction_&_Downstream.Rmd
Untracked: analysis/Cell_cycle_regression.Rmd
Untracked: analysis/Clustering_Tonsils_v2.Rmd
Untracked: analysis/DGE_earlyAIR_tAtlas_CD4_TCM.Rmd
Untracked: analysis/DGE_earlyAIR_tAtlas_CD4_TFH.Rmd
Untracked: analysis/DGE_earlyAIR_tAtlas_Naïve_Bcells.Rmd
Untracked: analysis/DGE_earlyAIR_vs_TonsilAtlas.Rmd
Untracked: analysis/Master_metadata.Rmd
Untracked: analysis/Pediatric_Vs_Adult_Atlases.Rmd
Untracked: analysis/Preprocessing_Batch1_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch2_Tonsils.Rmd
Untracked: analysis/Preprocessing_Batch3_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch4_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch5_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch6_BAL.Rmd
Untracked: analysis/Preprocessing_Batch7_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch8_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch9_Tonsils.Rmd
Untracked: analysis/TonsilsVsAdenoids.Rmd
Untracked: analysis/cell_cycle_regression.R
Untracked: analysis/testing_age_all.Rmd
Untracked: code/DGE_utils.R
Untracked: code/pseudobulk_analysis.R
Untracked: data/Cell_labels_Gunjan_v2/
Untracked: data/Cell_labels_Mel/
Untracked: data/Cell_labels_Mel_v2/
Untracked: data/Cell_labels_Mel_v3/
Untracked: data/Cell_labels_modified_Gunjan/
Untracked: data/Gene_sets/
Untracked: data/Hs.c2.cp.reactome.v7.1.entrez.rds
Untracked: data/Raw_feature_bc_matrix/
Untracked: data/cell_labels_Mel_v4_Dec2024/
Untracked: data/celltypes_Mel_GD_v3.xlsx
Untracked: data/celltypes_Mel_GD_v4_no_dups.xlsx
Untracked: data/celltypes_Mel_modified.xlsx
Untracked: data/celltypes_Mel_v2.csv
Untracked: data/celltypes_Mel_v2.xlsx
Untracked: data/celltypes_Mel_v2_MN.xlsx
Untracked: data/celltypes_for_mel_MN.xlsx
Untracked: data/col_palette.xlsx
Untracked: data/earlyAIR_sample_sheets_combined.xlsx
Untracked: figure/DGE_Tonsils_CD8.Rmd/
Untracked: output/CSV/All_tissues.propeller.xlsx
Untracked: output/CSV/Bronchial_brushings/
Untracked: output/CSV/Bronchial_brushings_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/
Untracked: output/CSV/G000231_Neeland_Adenoids.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Bronchial_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Nasal_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Tonsils.propeller.xlsx
Untracked: output/CSV/Nasal_brushings/
Untracked: output/DGE/Adenoids_B_memory/
Untracked: output/DGE/Adenoids_CD4/
Untracked: output/DGE/Adenoids_CD8/
Untracked: output/DGE/Adenoids_Cycling_GCB/
Untracked: output/DGE/Adenoids_IFN/
Untracked: output/DGE/RUV/
Untracked: output/DGE/RUVseq_TonsilAtlas/
Untracked: output/DGE/RUVseq_earlyAIR_Tonsils/
Untracked: output/DGE/TonsilAtlas/
Untracked: output/DGE/Tonsils/DGE_Age_summary_Tonsils.xlsx
Untracked: output/DGE/Tonsils/RUVTonsils_Memory B cells/
Untracked: output/DGE/Tonsils/RUV_Tonsils_Memory B cells/
Untracked: output/DGE/Tonsils/Tonsils_Naïve B cells/
Untracked: output/DGE/Tonsils_CD4/
Untracked: output/DGE/Tonsils_CD8/
Untracked: output/DGE/Tonsils_IFN/
Untracked: test_col.csv
Untracked: test_col.txt
Untracked: test_col.xlsx
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Modified: analysis/00_AllBatches_overview.Rmd
Modified: analysis/01_QC_emptyDrops.Rmd
Modified: analysis/02_QC_exploratoryPlots.Rmd
Modified: analysis/Adenoids.Rmd
Modified: analysis/Adenoids_v2.Rmd
Modified: analysis/Age_modeling.Rmd
Modified: analysis/Age_modelling_Adenoids.Rmd
Modified: analysis/AllBatches_QCExploratory.Rmd
Modified: analysis/BAL.Rmd
Modified: analysis/BAL_v2.Rmd
Modified: analysis/Bronchial_brushings.Rmd
Modified: analysis/Bronchial_brushings_v2.Rmd
Modified: analysis/DGE_TonsilAtlas.Rmd
Modified: analysis/DGE_Tonsils_CD8.Rmd
Modified: analysis/DGE_analysis_George.Rmd
Modified: analysis/Nasal_brushings.Rmd
Modified: analysis/Nasal_brushings_v2.Rmd
Modified: analysis/Subclustering_Adenoids.Rmd
Modified: analysis/Subclustering_BAL.Rmd
Modified: analysis/Subclustering_Bronchial_brushings.Rmd
Modified: analysis/Subclustering_Nasal_brushings.Rmd
Modified: analysis/Subclustering_Tonsils.Rmd
Modified: analysis/Tonsils.Rmd
Modified: analysis/Tonsils_v2.Rmd
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c9.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c9.csv
Modified: output/CSV_v2/G000231_Neeland_Nasal_brushings.propeller.xlsx
Staged changes:
New: test
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
suppressPackageStartupMessages({
library(here)
library(glue)
library(patchwork)
library(Seurat)
library(dplyr)
library(tidyverse)
library(gridExtra)
library(paletteer)
library(viridis)
library(tidyverse)
library(scran)
library(scater)
library(ggridges)
library(speckle)
library(edgeR)
library(limma)
library(knitr)
library(BiocStyle)
library(org.Hs.eg.db)
library(Glimma)
library(RUVSeq)
library(ggvenn)
library(writexl)
})
source(here("code/DGE_utils.R"))data_path <- here("output/RDS/AllBatches_Annotation_SEUs_v2/")
tissue <- "Tonsils"
seu <- readRDS(paste0(data_path, "G000231_Neeland_", tissue, ".annotated_clusters.SEU.rds"))seu <- JoinLayers(seu)Convert from Seurat to SingleCellExperiment object
sce_earlyAIR <- SingleCellExperiment(list(counts = seu@assays$RNA@layers$counts),
colData = seu@meta.data)
rownames(sce_earlyAIR) <- rownames(seu)seu_tonsilAtlas <- readRDS(here("output/RDS/Other_Atlas_SEUs/Tonsil_Atlas_3P_SEU.rds"))
sce_tonsilAtlas <- SingleCellExperiment(list(counts = seu_tonsilAtlas@assays$RNA@layers$counts),
colData = seu_tonsilAtlas@meta.data)Loading required package: HDF5ArrayLoading required package: DelayedArrayLoading required package: Matrix
Attaching package: 'Matrix'The following object is masked from 'package:S4Vectors':
expandThe following objects are masked from 'package:tidyr':
expand, pack, unpackLoading required package: S4ArraysLoading required package: abind
Attaching package: 'S4Arrays'The following object is masked from 'package:abind':
abindThe following object is masked from 'package:base':
rowsumLoading required package: SparseArray
Attaching package: 'DelayedArray'The following object is masked from 'package:purrr':
simplifyThe following objects are masked from 'package:base':
apply, scale, sweepLoading required package: rhdf5
Attaching package: 'HDF5Array'The following object is masked from 'package:rhdf5':
h5lsrownames(sce_tonsilAtlas) <- rownames(seu_tonsilAtlas)camera gene-set testing
library(msigdbr)
msigdb <- msigdbr(species = "Homo sapiens")
development_sets <- msigdb %>% filter(gs_name %in% c("GO_EMBRYONIC_DEVELOPMENT", "GO_HEMATOPOIETIC_ORGAN_DEVELOPMENT", "REACTOME_DEVELOPMENTAL_BIOLOGY"))Make gene-set lists from files
Hs.c2.all <- convert_gmt_to_list(here("data/Gene_sets/c2.all.v2024.1.Hs.entrez.gmt"))
Hs.h.all <- convert_gmt_to_list(here("data/Gene_sets/h.all.v2024.1.Hs.entrez.gmt"))
Hs.c5.all <- convert_gmt_to_list(here("data/Gene_sets/c5.all.v2024.1.Hs.entrez.gmt"))
gene_sets_list <- list(HALLMARK = Hs.h.all,
GO = Hs.c5.all,
REACTOME = Hs.c2.all[str_detect(names(Hs.c2.all), "REACTOME")],
WP = Hs.c2.all[str_detect(names(Hs.c2.all), "^WP")])celltype <- "CD4 TCM"
dge_eAIR <- make_pb_earlyAIR(sce = sce_earlyAIR, celltype = celltype, sample_info = "sample_id",k = 1)Warning in RUVg(dge$counts, ctl, k = k): The expression matrix does not contain counts.
Please, pass a matrix of counts (not logged) or set isLog to TRUE to skip the log transformationfit_eAIR <- dge_eAIR@fit
lrt_eAIR <- dge_eAIR@lrtresults <- as.data.frame(topTags(lrt_eAIR,n = 50))
plotMD(lrt_eAIR, main = "Mean Difference: Age (earlyAIR)")
text(results$logCPM,results$logFC,labels = rownames(results),col="black",cex=0.5,pos=3)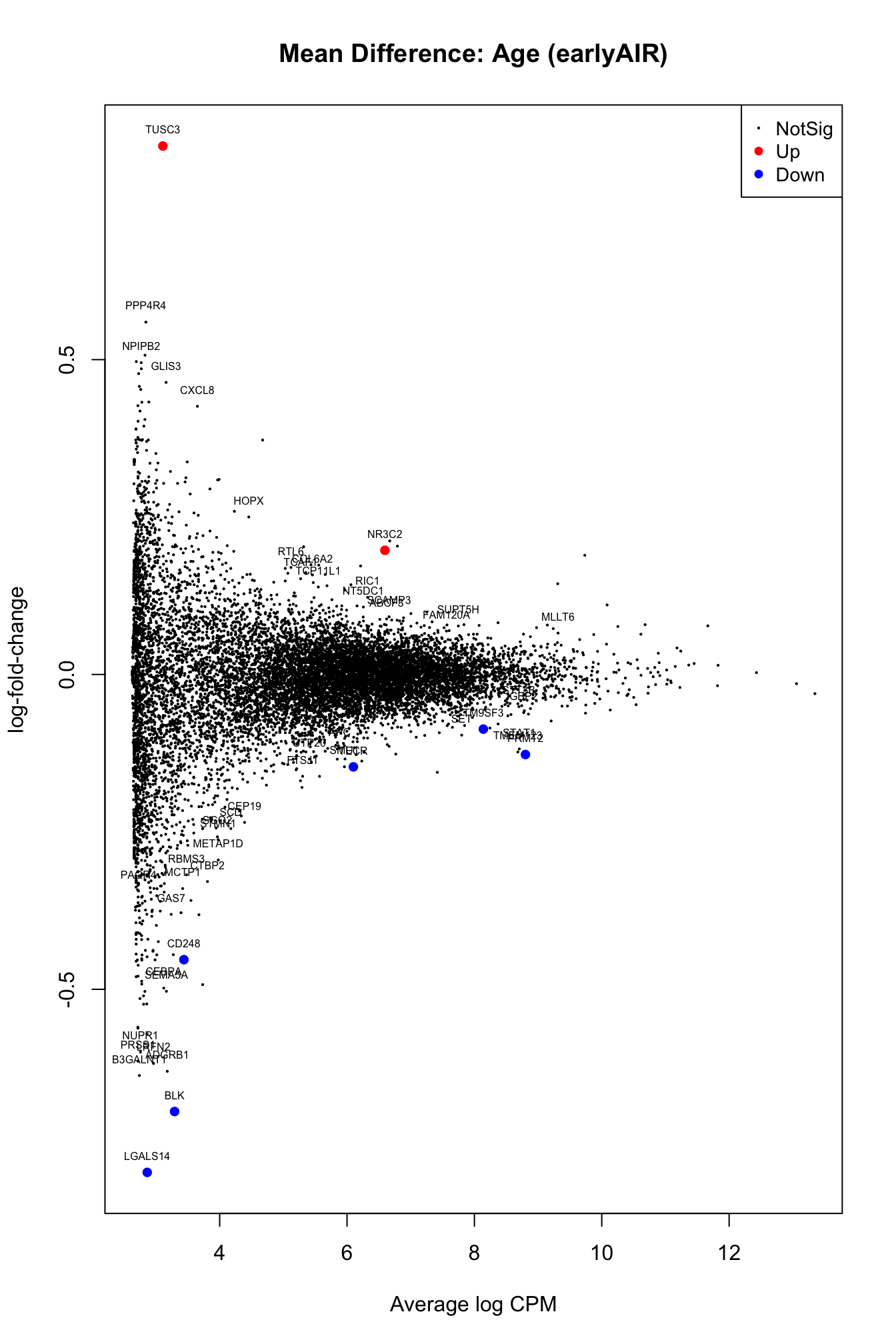
library(org.Hs.eg.db)
gns <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = rownames(fit_eAIR),
column = c("ENTREZID"),
keytype = "SYMBOL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnsoutput_path <- here("output/DGE/RUV", tissue)
output_dir <- file.path(output_path, glue("RUV_earlyAIR_{celltype}"))
dir.create(output_dir, recursive = TRUE, showWarnings = FALSE)
t.stat <- sign(lrt_eAIR$table$logFC) * sqrt(lrt_eAIR$table$LR) %>% na.omit()Warning in sqrt(lrt_eAIR$table$LR): NaNs producedWarning in sign(lrt_eAIR$table$logFC) * sqrt(lrt_eAIR$table$LR) %>% na.omit():
longer object length is not a multiple of shorter object lengthtest_set <- gene_set_test_camera(gene_sets_list, gns, fit_eAIR, t.stat, cellDir = output_dir)p1 <- top_camera_sets(test_set, num = 10) + ggtitle(paste0(celltype, " :earlyAIR"))dge_tATLAS <- make_pb_earlyAIR(sce = sce_tonsilAtlas, celltype = celltype, sample_info = "donor_id",k = 2)
fit_tATLAS <- dge_tATLAS@fit
lrt_tATLAS <- dge_tATLAS@lrtresults <- as.data.frame(topTags(lrt_tATLAS,n = 50))
plotMD(lrt_tATLAS, main = "Mean Difference: Age (tonsilATLAS)")
text(results$logCPM,results$logFC,labels = rownames(results),col="black",cex=0.5,pos=3)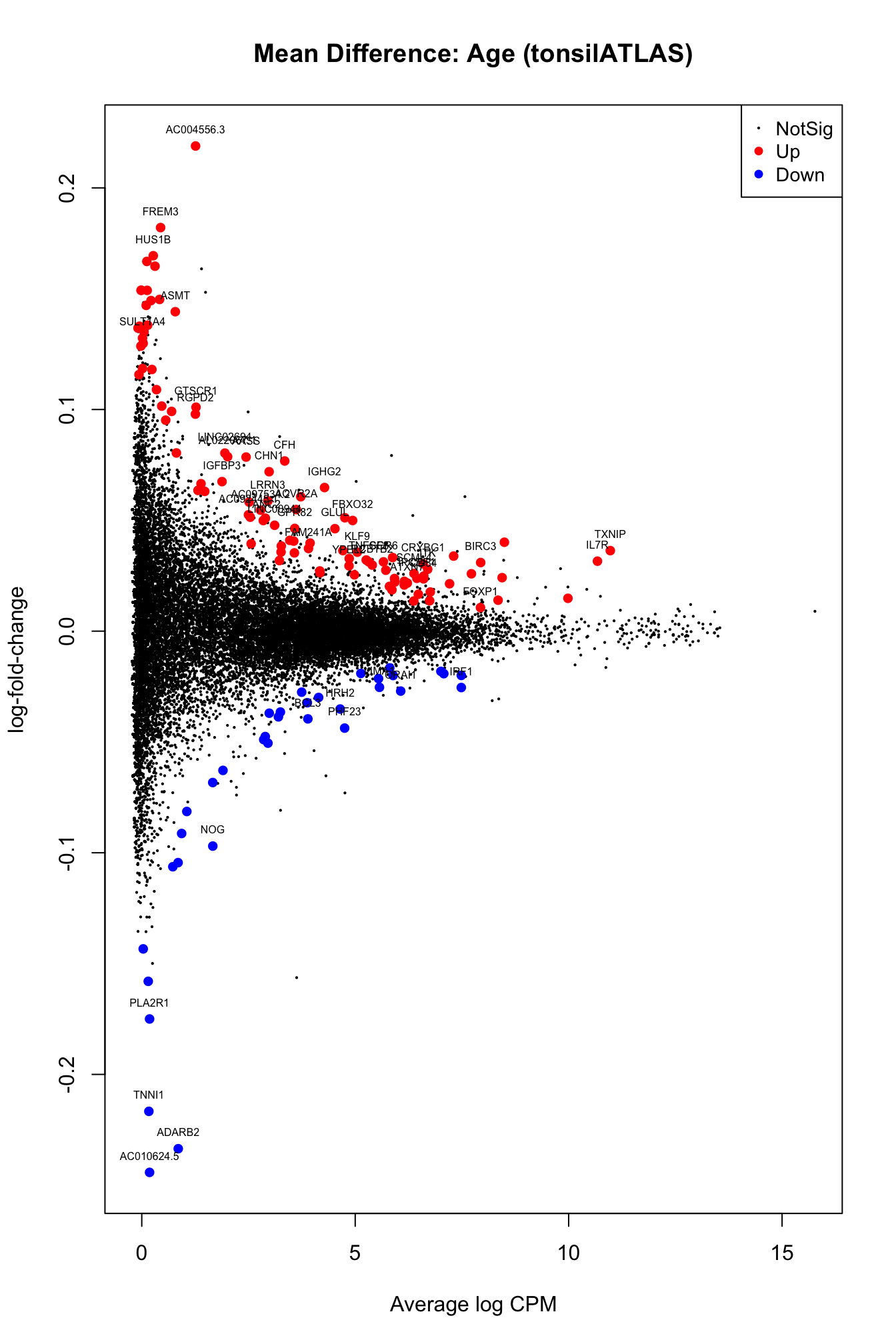
library(org.Hs.eg.db)
gns <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = rownames(fit_tATLAS),
column = c("ENTREZID"),
keytype = "SYMBOL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnsoutput_path <- here("output/DGE/RUV", tissue)
output_dir <- file.path(output_path, glue("RUV_TonsilATLAS_{celltype}"))
dir.create(output_dir, recursive = TRUE, showWarnings = FALSE)
t.stat_tA <- sign(lrt_tATLAS$table$logFC) * sqrt(lrt_tATLAS$table$LR) %>% na.omit()Warning in sqrt(lrt_tATLAS$table$LR): NaNs producedWarning in sign(lrt_tATLAS$table$logFC) * sqrt(lrt_tATLAS$table$LR) %>% :
longer object length is not a multiple of shorter object lengthtest_set_tA <- gene_set_test_camera(gene_sets_list, gns, fit_tATLAS, t.stat_tA, cellDir = output_dir)p2 <- top_camera_sets(test_set_tA, num = 10) + ggtitle(paste0(celltype, " :TonsilAtlas"))p1 / p2 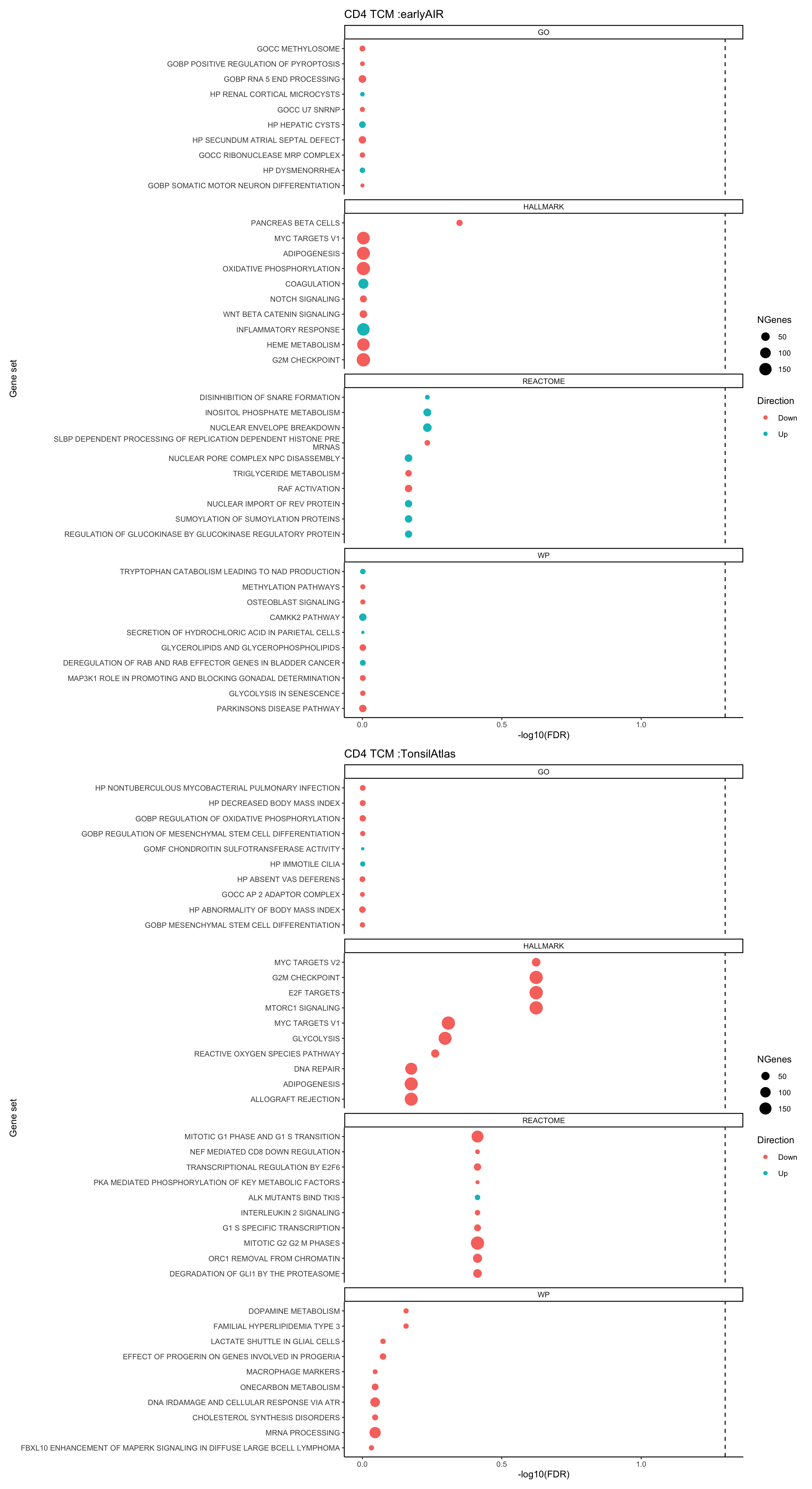
earlyAIR_top <- rownames(topTags(lrt_eAIR, n=Inf))
tonsilAtlas_top <- rownames(topTags(lrt_tATLAS, n=Inf))
venn_data <- list(
earlyAIR = earlyAIR_top,
tonsilAtlas = tonsilAtlas_top
)
ggvenn(venn_data, fill_color = c("orange", "hotpink")) +
ggtitle(paste(celltype))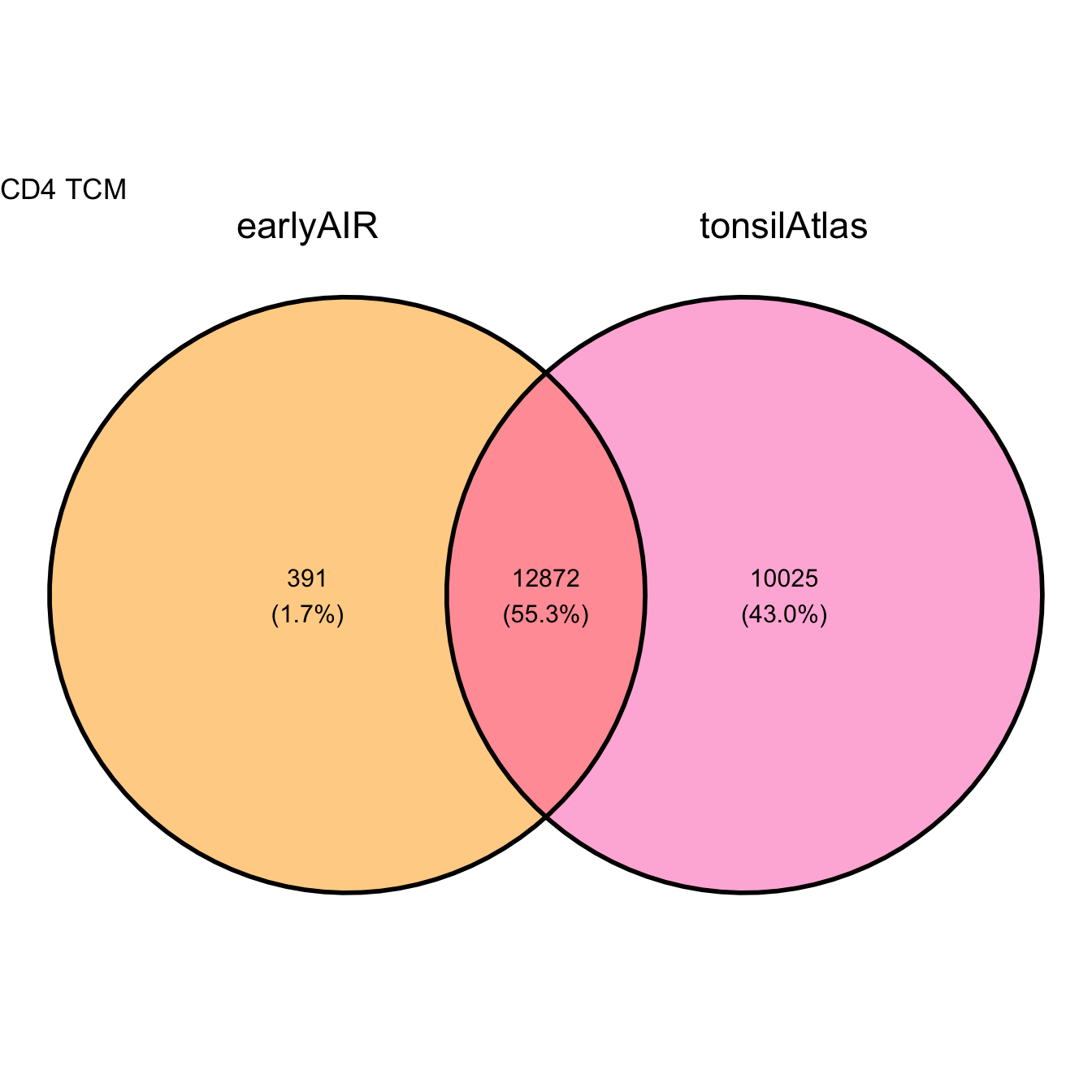
unique_earlyAIR <- setdiff(earlyAIR_top, tonsilAtlas_top)
unique_TonsilAtlas <- setdiff(tonsilAtlas_top, earlyAIR_top)
common_DEGs <- intersect(earlyAIR_top, tonsilAtlas_top)library(clusterProfiler)
library(ReactomePA)
# Convert gene symbols to Entrez IDs
gns_unique_earlyAIR <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = unique_earlyAIR,
column = "ENTREZID",
keytype = "SYMBOL",
multiVals = "first")
# Run GO analysis (Biological Process)
go_results <- enrichGO(gene = gns_unique_earlyAIR,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pAdjustMethod = "BH",
readable = TRUE)library(ggplot2)
library(enrichplot)
# Barplot for GO enrichment
barplot(go_results, showCategory = 20) + ggtitle("GO Enrichment - EarlyAIR Unique Genes")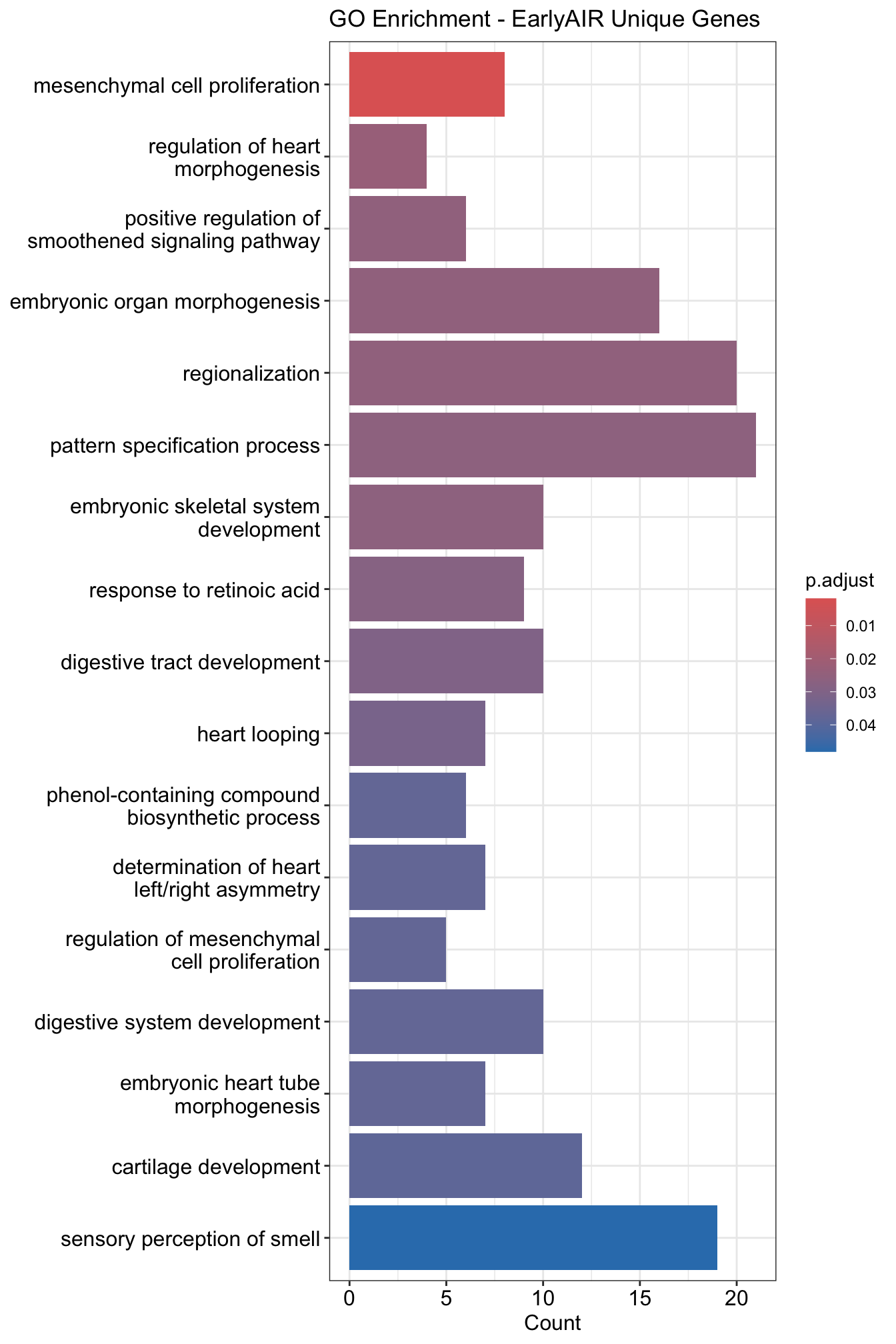
# Dotplot for KEGG pathways
#dotplot(kegg_results, showCategory = 20) + ggtitle("KEGG Pathways - EarlyAIR Unique Genes")
# Dotplot for Reactome pathways
#dotplot(reactome_results, showCategory = 20) + ggtitle("Reactome Pathways - EarlyAIR Unique Genes")gns_unique_TonsilAtlas <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = unique_TonsilAtlas,
column = "ENTREZID",
keytype = "SYMBOL",
multiVals = "first")'select()' returned 1:1 mapping between keys and columnsgns_unique_TonsilAtlas <- na.omit(gns_unique_TonsilAtlas) # Remove NAs
go_results_TonsilAtlas <- enrichGO(gene = gns_unique_TonsilAtlas,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pAdjustMethod = "BH",
readable = TRUE)
# Extract top pathways
go_df_earlyAIR <- go_results@result %>%
filter(p.adjust < 0.05) %>%
mutate(Group = "earlyAIR") %>%
arrange(p.adjust) %>%
head(50)
go_df_TonsilAtlas <- go_results_TonsilAtlas@result %>%
filter(p.adjust < 0.05) %>%
mutate(Group = "TonsilAtlas") %>%
arrange(p.adjust) %>%
head(50)library(ggplot2)
library(forcats)
# Function to plot pathways
plot_go_enrichment <- function(go_df, title, fill_color) {
ggplot(go_df, aes(x = -log10(p.adjust), y = fct_reorder(Description, -p.adjust), fill = Group)) +
geom_col() +
scale_fill_manual(values = fill_color) +
labs(title = title, x = "-log10(p.adjust)", y = "GO Term") +
theme_minimal()
}
p1 <- plot_go_enrichment(go_df_earlyAIR, "Top GO Pathways - earlyAIR", "orange")
p2 <- plot_go_enrichment(go_df_TonsilAtlas, "Top GO Pathways - TonsilAtlas", "purple")p1 / p2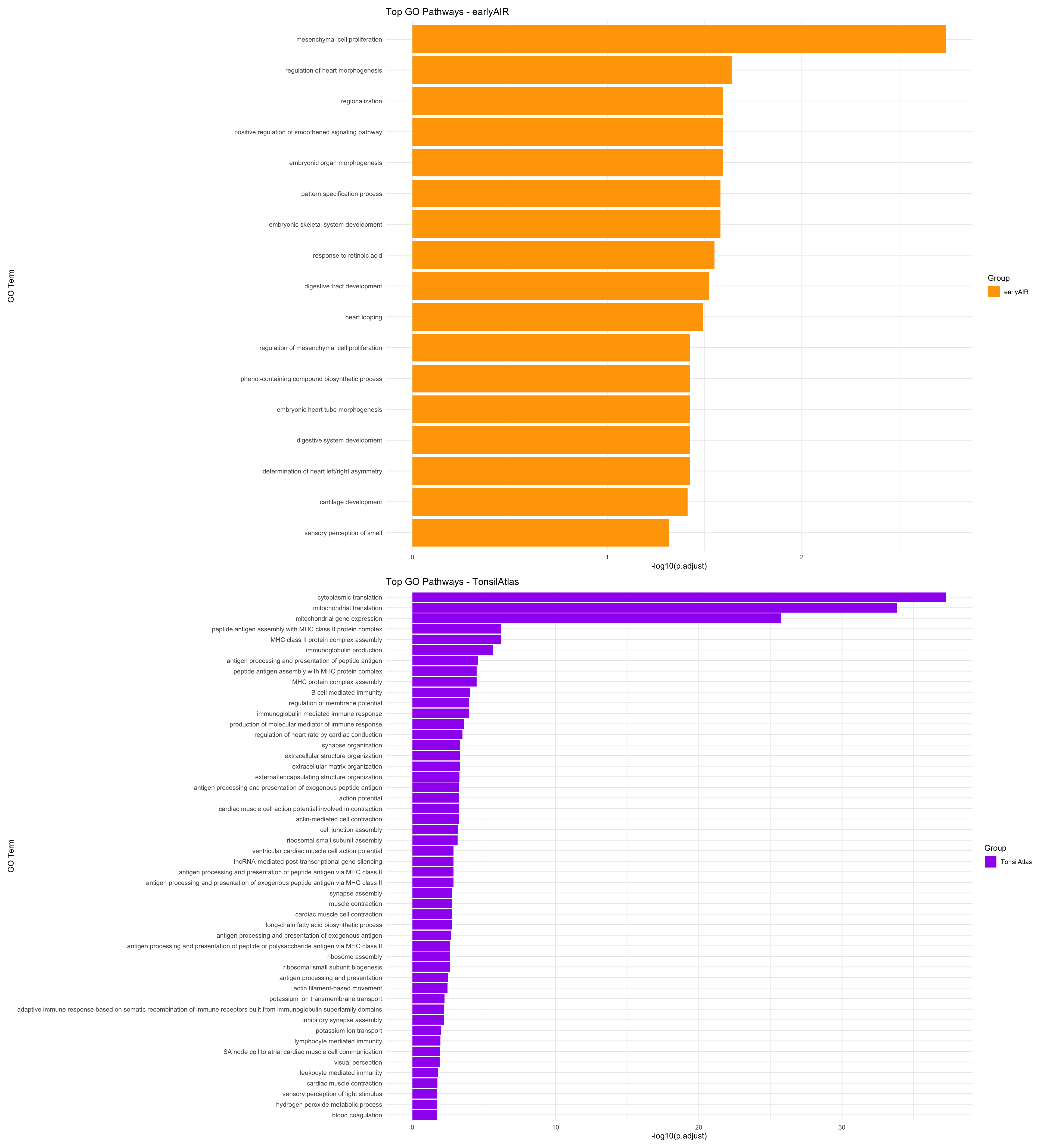
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.3.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] enrichplot_1.22.0 ReactomePA_1.46.0
[3] clusterProfiler_4.10.1 msigdbr_7.5.1
[5] HDF5Array_1.30.1 rhdf5_2.46.1
[7] DelayedArray_0.28.0 SparseArray_1.2.4
[9] S4Arrays_1.2.0 abind_1.4-5
[11] Matrix_1.6-5 scMerge_1.18.0
[13] writexl_1.5.0 ggvenn_0.1.10
[15] RUVSeq_1.36.0 EDASeq_2.36.0
[17] ShortRead_1.60.0 GenomicAlignments_1.38.2
[19] Rsamtools_2.18.0 Biostrings_2.70.2
[21] XVector_0.42.0 BiocParallel_1.36.0
[23] Glimma_2.12.0 org.Hs.eg.db_3.18.0
[25] AnnotationDbi_1.64.1 BiocStyle_2.30.0
[27] knitr_1.45 edgeR_4.0.16
[29] limma_3.58.1 speckle_1.2.0
[31] ggridges_0.5.6 scater_1.30.1
[33] scran_1.30.2 scuttle_1.12.0
[35] SingleCellExperiment_1.24.0 SummarizedExperiment_1.32.0
[37] Biobase_2.62.0 GenomicRanges_1.54.1
[39] GenomeInfoDb_1.38.6 IRanges_2.36.0
[41] S4Vectors_0.40.2 BiocGenerics_0.48.1
[43] MatrixGenerics_1.14.0 matrixStats_1.2.0
[45] viridis_0.6.5 viridisLite_0.4.2
[47] paletteer_1.6.0 gridExtra_2.3
[49] lubridate_1.9.3 forcats_1.0.0
[51] stringr_1.5.1 purrr_1.0.2
[53] readr_2.1.5 tidyr_1.3.1
[55] tibble_3.2.1 ggplot2_3.5.0
[57] tidyverse_2.0.0 dplyr_1.1.4
[59] Seurat_5.0.1.9009 SeuratObject_5.0.1
[61] sp_2.1-3 patchwork_1.2.0
[63] glue_1.7.0 here_1.0.1
[65] workflowr_1.7.1
loaded via a namespace (and not attached):
[1] graph_1.80.0 igraph_2.0.2
[3] ica_1.0-3 plotly_4.10.4
[5] Formula_1.2-5 rematch2_2.1.2
[7] zlibbioc_1.48.0 tidyselect_1.2.0
[9] bit_4.0.5 lattice_0.22-5
[11] rjson_0.2.21 M3Drop_1.28.0
[13] blob_1.2.4 parallel_4.3.2
[15] png_0.1-8 ResidualMatrix_1.12.0
[17] ggplotify_0.1.2 cli_3.6.2
[19] goftest_1.2-3 BiocIO_1.12.0
[21] bluster_1.12.0 BiocNeighbors_1.20.2
[23] densEstBayes_1.0-2.2 shadowtext_0.1.3
[25] uwot_0.1.16 curl_5.2.0
[27] tidytree_0.4.6 mime_0.12
[29] evaluate_0.23 leiden_0.4.3.1
[31] stringi_1.8.3 backports_1.4.1
[33] XML_3.99-0.16.1 httpuv_1.6.14
[35] magrittr_2.0.3 rappdirs_0.3.3
[37] splines_4.3.2 jpeg_0.1-10
[39] ggraph_2.1.0 sctransform_0.4.1
[41] ggbeeswarm_0.7.2 DBI_1.2.2
[43] reactome.db_1.86.2 jquerylib_0.1.4
[45] withr_3.0.0 git2r_0.33.0
[47] rprojroot_2.0.4 lmtest_0.9-40
[49] tidygraph_1.3.1 bdsmatrix_1.3-6
[51] rtracklayer_1.62.0 BiocManager_1.30.22
[53] htmlwidgets_1.6.4 fs_1.6.3
[55] biomaRt_2.58.2 ggrepel_0.9.5
[57] labeling_0.4.3 DESeq2_1.42.1
[59] DEoptimR_1.1-3 reticulate_1.35.0
[61] zoo_1.8-12 timechange_0.3.0
[63] fansi_1.0.6 caTools_1.18.2
[65] ggtree_3.10.1 data.table_1.15.0
[67] ruv_0.9.7.1 R.oo_1.26.0
[69] RSpectra_0.16-1 irlba_2.3.5.1
[71] gridGraphics_0.5-1 fastDummies_1.7.3
[73] ellipsis_0.3.2 aroma.light_3.32.0
[75] lazyeval_0.2.2 yaml_2.3.8
[77] survival_3.5-8 scattermore_1.2
[79] crayon_1.5.2 RcppAnnoy_0.0.22
[81] tweenr_2.0.3 RColorBrewer_1.1-3
[83] progressr_0.14.0 later_1.3.2
[85] codetools_0.2-19 base64enc_0.1-3
[87] KEGGREST_1.42.0 bbmle_1.0.25.1
[89] Rtsne_0.17 startupmsg_0.9.6.1
[91] filelock_1.0.3 foreign_0.8-86
[93] pkgconfig_2.0.3 xml2_1.3.6
[95] getPass_0.2-4 sfsmisc_1.1-17
[97] aplot_0.2.2 ape_5.8
[99] spatstat.sparse_3.0-3 xtable_1.8-4
[101] interp_1.1-6 hwriter_1.3.2.1
[103] highr_0.10 plyr_1.8.9
[105] httr_1.4.7 tools_4.3.2
[107] globals_0.16.2 pkgbuild_1.4.3
[109] beeswarm_0.4.0 htmlTable_2.4.2
[111] checkmate_2.3.1 nlme_3.1-164
[113] loo_2.7.0 HDO.db_0.99.1
[115] dbplyr_2.4.0 digest_0.6.34
[117] numDeriv_2016.8-1.1 farver_2.1.1
[119] tzdb_0.4.0 reshape2_1.4.4
[121] yulab.utils_0.1.8 cvTools_0.3.2
[123] rpart_4.1.23 cachem_1.0.8
[125] BiocFileCache_2.10.1 polyclip_1.10-6
[127] Hmisc_5.1-1 generics_0.1.3
[129] proxyC_0.3.4 mvtnorm_1.2-4
[131] parallelly_1.37.0 statmod_1.5.0
[133] RcppHNSW_0.6.0 ScaledMatrix_1.10.0
[135] pbapply_1.7-2 spam_2.10-0
[137] gson_0.1.0 dqrng_0.3.2
[139] utf8_1.2.4 graphlayouts_1.1.0
[141] StanHeaders_2.32.5 gtools_3.9.5
[143] shiny_1.8.0 GenomeInfoDbData_1.2.11
[145] R.utils_2.12.3 rhdf5filters_1.14.1
[147] RCurl_1.98-1.14 memoise_2.0.1
[149] rmarkdown_2.25 scales_1.3.0
[151] R.methodsS3_1.8.2 future_1.33.1
[153] RANN_2.6.1 spatstat.data_3.0-4
[155] rstudioapi_0.15.0 cluster_2.1.6
[157] QuickJSR_1.1.3 whisker_0.4.1
[159] rstantools_2.4.0 spatstat.utils_3.0-4
[161] hms_1.1.3 fitdistrplus_1.1-11
[163] munsell_0.5.0 cowplot_1.1.3
[165] colorspace_2.1-0 rlang_1.1.3
[167] DelayedMatrixStats_1.24.0 sparseMatrixStats_1.14.0
[169] dotCall64_1.1-1 ggforce_0.4.2
[171] mgcv_1.9-1 xfun_0.42
[173] reldist_1.7-2 GOSemSim_2.28.1
[175] rstan_2.32.5 treeio_1.26.0
[177] Rhdf5lib_1.24.2 bitops_1.0-7
[179] ps_1.7.6 promises_1.2.1
[181] scatterpie_0.2.4 inline_0.3.19
[183] RSQLite_2.3.5 qvalue_2.34.0
[185] fgsea_1.28.0 GO.db_3.18.0
[187] compiler_4.3.2 prettyunits_1.2.0
[189] beachmat_2.18.1 graphite_1.48.0
[191] listenv_0.9.1 Rcpp_1.0.12
[193] BiocSingular_1.18.0 tensor_1.5
[195] MASS_7.3-60.0.1 progress_1.2.3
[197] babelgene_22.9 spatstat.random_3.2-2
[199] R6_2.5.1 fastmap_1.1.1
[201] fastmatch_1.1-4 vipor_0.4.7
[203] distr_2.9.3 ROCR_1.0-11
[205] rsvd_1.0.5 nnet_7.3-19
[207] gtable_0.3.4 KernSmooth_2.23-22
[209] latticeExtra_0.6-30 miniUI_0.1.1.1
[211] deldir_2.0-2 htmltools_0.5.7
[213] RcppParallel_5.1.7 bit64_4.0.5
[215] spatstat.explore_3.2-6 lifecycle_1.0.4
[217] processx_3.8.3 callr_3.7.5
[219] restfulr_0.0.15 sass_0.4.8
[221] vctrs_0.6.5 spatstat.geom_3.2-8
[223] robustbase_0.99-2 DOSE_3.28.2
[225] ggfun_0.1.4 future.apply_1.11.1
[227] bslib_0.6.1 pillar_1.9.0
[229] batchelor_1.18.1 GenomicFeatures_1.54.3
[231] gplots_3.1.3.1 metapod_1.10.1
[233] locfit_1.5-9.8 jsonlite_1.8.8