Nasal_brushings_v2
Clustering and Marker gene analysis
Gunjan Dixit
January 15, 2025
Last updated: 2025-01-15
Checks: 6 1
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 54e4ec2. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/RDS/
Ignored: output/.DS_Store
Ignored: output/CSV/.DS_Store
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Ignored: output/G000231_Neeland_batch9_1/
Ignored: output/RDS/
Ignored: output/plots/
Untracked files:
Untracked: All_Batches_QCExploratory_v2.Rmd
Untracked: Annotation_Bronchial_brushings.Rmd
Untracked: BAL_Tcell_propeller.xlsx
Untracked: BAL_propeller.xlsx
Untracked: BB_Tcell_propeller.xlsx
Untracked: BB_propeller.xlsx
Untracked: NB_Tcell_propeller.xlsx
Untracked: NB_propeller.csv
Untracked: NB_propeller.xlsx
Untracked: Tonsil_Atlas.SCE.rds
Untracked: analysis/03_Batch_Integration.Rmd
Untracked: analysis/Age_proportions.Rmd
Untracked: analysis/Age_proportions_AllBatches.Rmd
Untracked: analysis/Annotation_BAL.Rmd
Untracked: analysis/Annotation_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Adenoids.Rmd
Untracked: analysis/BatchCorrection_Nasal_brushings.Rmd
Untracked: analysis/BatchCorrection_Tonsils.Rmd
Untracked: analysis/Batch_Integration_&_Downstream_analysis.Rmd
Untracked: analysis/Batch_correction_&_Downstream.Rmd
Untracked: analysis/Cell_cycle_regression.Rmd
Untracked: analysis/Clustering_Tonsils_v2.Rmd
Untracked: analysis/Master_metadata.Rmd
Untracked: analysis/Pediatric_Vs_Adult_Atlases.Rmd
Untracked: analysis/Preprocessing_Batch1_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch2_Tonsils.Rmd
Untracked: analysis/Preprocessing_Batch3_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch4_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch5_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch6_BAL.Rmd
Untracked: analysis/Preprocessing_Batch7_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch8_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch9_Tonsils.Rmd
Untracked: analysis/TonsilsVsAdenoids.Rmd
Untracked: analysis/cell_cycle_regression.R
Untracked: analysis/testing_age_all.Rmd
Untracked: color_palette.rds
Untracked: color_palette_Oct_2024.rds
Untracked: color_palette_v2_level2.rds
Untracked: combined_metadata.rds
Untracked: data/Cell_labels_Gunjan_v2/
Untracked: data/Cell_labels_Mel/
Untracked: data/Cell_labels_Mel_v2/
Untracked: data/Cell_labels_Mel_v3/
Untracked: data/Cell_labels_modified_Gunjan/
Untracked: data/Hs.c2.cp.reactome.v7.1.entrez.rds
Untracked: data/Raw_feature_bc_matrix/
Untracked: data/cell_labels_Mel_v4_Dec2024/
Untracked: data/celltypes_Mel_GD_v3.xlsx
Untracked: data/celltypes_Mel_GD_v4_no_dups.xlsx
Untracked: data/celltypes_Mel_modified.xlsx
Untracked: data/celltypes_Mel_v2.csv
Untracked: data/celltypes_Mel_v2.xlsx
Untracked: data/celltypes_Mel_v2_MN.xlsx
Untracked: data/celltypes_for_mel_MN.xlsx
Untracked: data/earlyAIR_sample_sheets_combined.xlsx
Untracked: output/CSV/All_tissues.propeller.xlsx
Untracked: output/CSV/Bronchial_brushings/
Untracked: output/CSV/Bronchial_brushings_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/
Untracked: output/CSV/G000231_Neeland_Adenoids.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Bronchial_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Nasal_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Tonsils.propeller.xlsx
Untracked: output/CSV/Nasal_brushings/
Untracked: tonsil_atlas_metadata.png
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Modified: analysis/00_AllBatches_overview.Rmd
Modified: analysis/01_QC_emptyDrops.Rmd
Modified: analysis/02_QC_exploratoryPlots.Rmd
Modified: analysis/Adenoids.Rmd
Modified: analysis/Age_modeling.Rmd
Modified: analysis/Age_modelling_Adenoids.Rmd
Modified: analysis/Age_modelling_Tonsils.Rmd
Modified: analysis/AllBatches_QCExploratory.Rmd
Modified: analysis/BAL.Rmd
Modified: analysis/Bronchial_brushings.Rmd
Modified: analysis/Bronchial_brushings_v2.Rmd
Modified: analysis/Nasal_brushings.Rmd
Modified: analysis/Nasal_brushings_v2.Rmd
Modified: analysis/Subclustering_Adenoids.Rmd
Modified: analysis/Subclustering_BAL.Rmd
Modified: analysis/Subclustering_Bronchial_brushings.Rmd
Modified: analysis/Subclustering_Nasal_brushings.Rmd
Modified: analysis/Subclustering_Tonsils.Rmd
Modified: analysis/Tonsils.Rmd
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c9.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c9.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Nasal_brushings_v2.Rmd)
and HTML (docs/Nasal_brushings_v2.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 54e4ec2 | Gunjan Dixit | 2025-01-08 | updated clustering annotations |
| html | 54e4ec2 | Gunjan Dixit | 2025-01-08 | updated clustering annotations |
| Rmd | 3595ad0 | Gunjan Dixit | 2025-01-07 | Added B cell subclustering |
| html | 3595ad0 | Gunjan Dixit | 2025-01-07 | Added B cell subclustering |
| Rmd | eebc9b9 | Gunjan Dixit | 2024-12-22 | Updated NB, BB clustering |
| html | eebc9b9 | Gunjan Dixit | 2024-12-22 | Updated NB, BB clustering |
Introduction
This Rmarkdown file loads and analyzes the batch-integrated/merged
Seurat object for Nasal Brushings (Batch1 and Batch5).
It performs clustering at various resolutions ranging from 0-1, followed
by visualization of identified clusters and Broad Level 3 cell labels on
UMAP. Next, the FindAllMarkers function is used to perform
marker gene analysis to identify marker genes for each cluster. The top
marker gene is visualized using FeaturePlot,
ViolinPlot and Heatmap. The identified marker
genes are stored in CSV format for each cluster at the optimum
resolution identified using clustree function.
Load libraries
suppressPackageStartupMessages({
library(BiocStyle)
library(tidyverse)
library(here)
library(glue)
library(dplyr)
library(Seurat)
library(clustree)
library(kableExtra)
library(RColorBrewer)
library(data.table)
library(ggplot2)
library(patchwork)
library(limma)
library(edgeR)
library(speckle)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(readxl)
})Load Input data
Load merged object (batch corrected/integrated) for the tissue.
tissue <- "Nasal_brushings"
out <- here("output/RDS/AllBatches_Harmony_SEUs_v2/G000231_Neeland_Nasal_brushings_batchCorrection.Harmony.clusters.SEU.rds")
merged_obj <- readRDS(out)
merged_objAn object of class Seurat
17973 features across 93832 samples within 1 assay
Active assay: RNA (17973 features, 2000 variable features)
5 layers present: counts.G000231_batch1, counts.G000231_batch5, scale.data, data.G000231_batch1, data.G000231_batch5
4 dimensional reductions calculated: pca, umap.unintegrated, harmony, umap.harmonyClustering
Clustering is done on the “harmony” or batch integrated reduction at resolutions ranging from 0-1.
out1 <- here("output",
"RDS", "AllBatches_Clustering_SEUs_v2",
paste0("G000231_Neeland_",tissue,".Clusters.SEU.rds"))
#dir.create(out1)
resolutions <- seq(0.1, 1, by = 0.1)
if (!file.exists(out1)) {
merged_obj <- FindNeighbors(merged_obj, reduction = "harmony", dims = 1:30)
merged_obj <- FindClusters(merged_obj, resolution = seq(0.1, 1, by = 0.1), algorithm = 3)
saveRDS(merged_obj, file = out1)
} else {
merged_obj <- readRDS(out1)
}The clustree function is used to visualize the
clustering at different resolutions to identify the most optimum
resolution.
clustree(merged_obj, prefix = "RNA_snn_res.")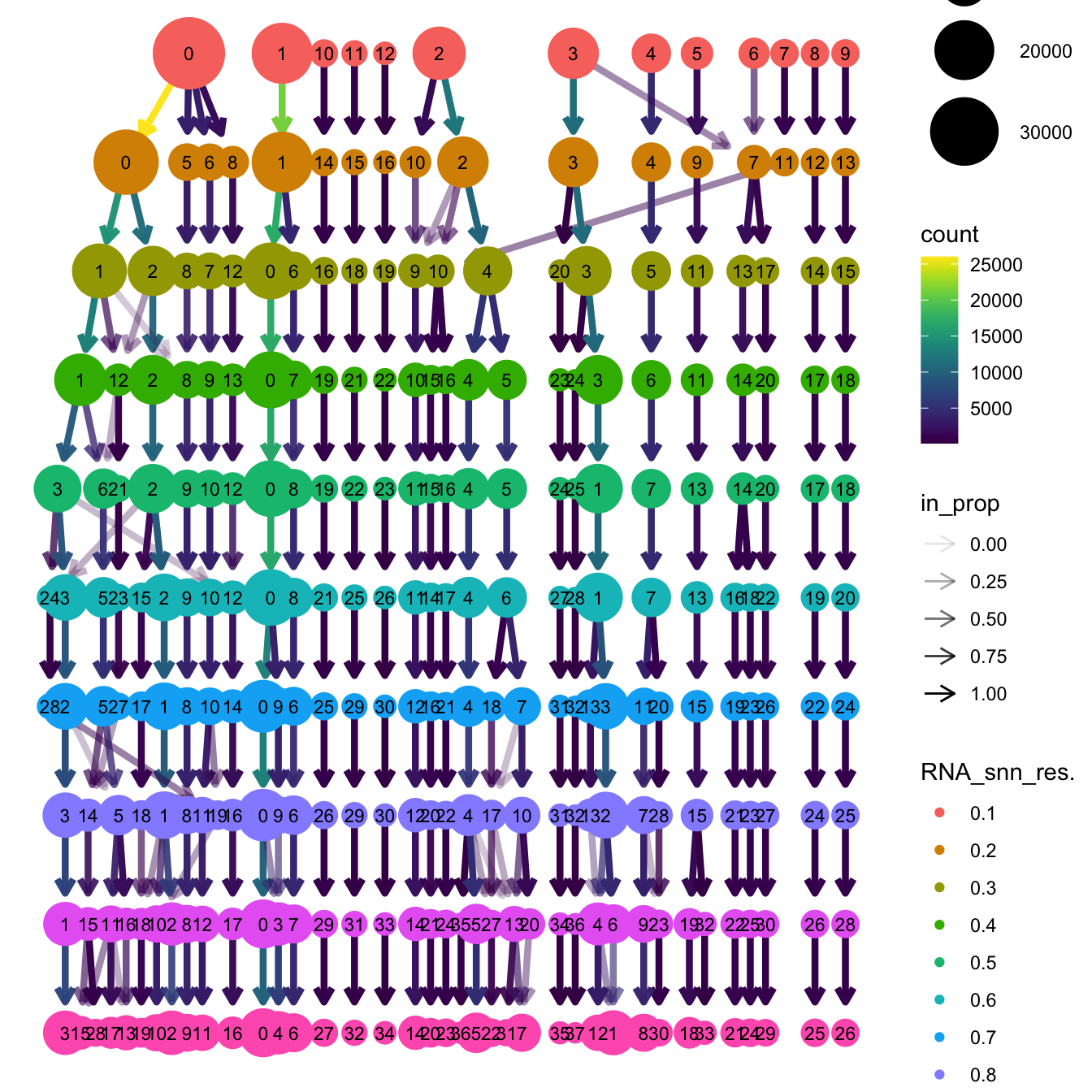
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Based on the clustering tree, we chose an intermediate/optimum resolution of 0.4 where the clustering results are the most stable, with the least amount of shuffling cells.
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(merged_obj$RNA_snn_res.0.4)
merged_obj$RNA_snn_res.0.4 <- factor(merged_obj$RNA_snn_res.0.4, levels = seq(0,n-1))
merged_obj$seurat_clusters <- NULL
merged_obj$cluster <- merged_obj$RNA_snn_res.0.4
Idents(merged_obj) <- merged_obj$clusterUMAP after clustering
Defining colours for each cell-type to be consistent with other age-related/cell type composition plots.
my_colors <- c(
"B cells" = "steelblue",
"CD4 T cells" = "brown",
"Double negative T cells" = "gold",
"CD8 T cells" = "lightgreen",
"Pre B/T cells" = "orchid",
"Innate lymphoid cells" = "tan",
"Natural Killer cells" = "blueviolet",
"Macrophages" = "green4",
"Cycling T cells" = "turquoise",
"Dendritic cells" = "grey80",
"Gamma delta T cells" = "mediumvioletred",
"Epithelial lineage" = "darkorange",
"Granulocytes" = "olivedrab",
"Fibroblast lineage" = "lavender",
"None" = "white",
"Monocytes" = "peachpuff",
"Endothelial lineage" = "cadetblue",
"SMG duct" = "lightpink",
"Neuroendocrine" = "skyblue",
"Doublet query/Other" = "#d62728"
)UMAP displaying clusters at opt_res resolution and Broad
cell Labels Level 3.
p1 <- DimPlot(merged_obj, reduction = "umap.harmony", raster = FALSE ,repel = TRUE, label = TRUE,label.size = 3.5, group.by = opt_res) + NoLegend()
p2 <- DimPlot(merged_obj, reduction = "umap.harmony", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5, group.by = "Broad_cell_label_3") +
scale_colour_manual(values = my_colors) +
ggtitle(paste0(tissue, ": Batch Corrected UMAP"))
p1 / p2 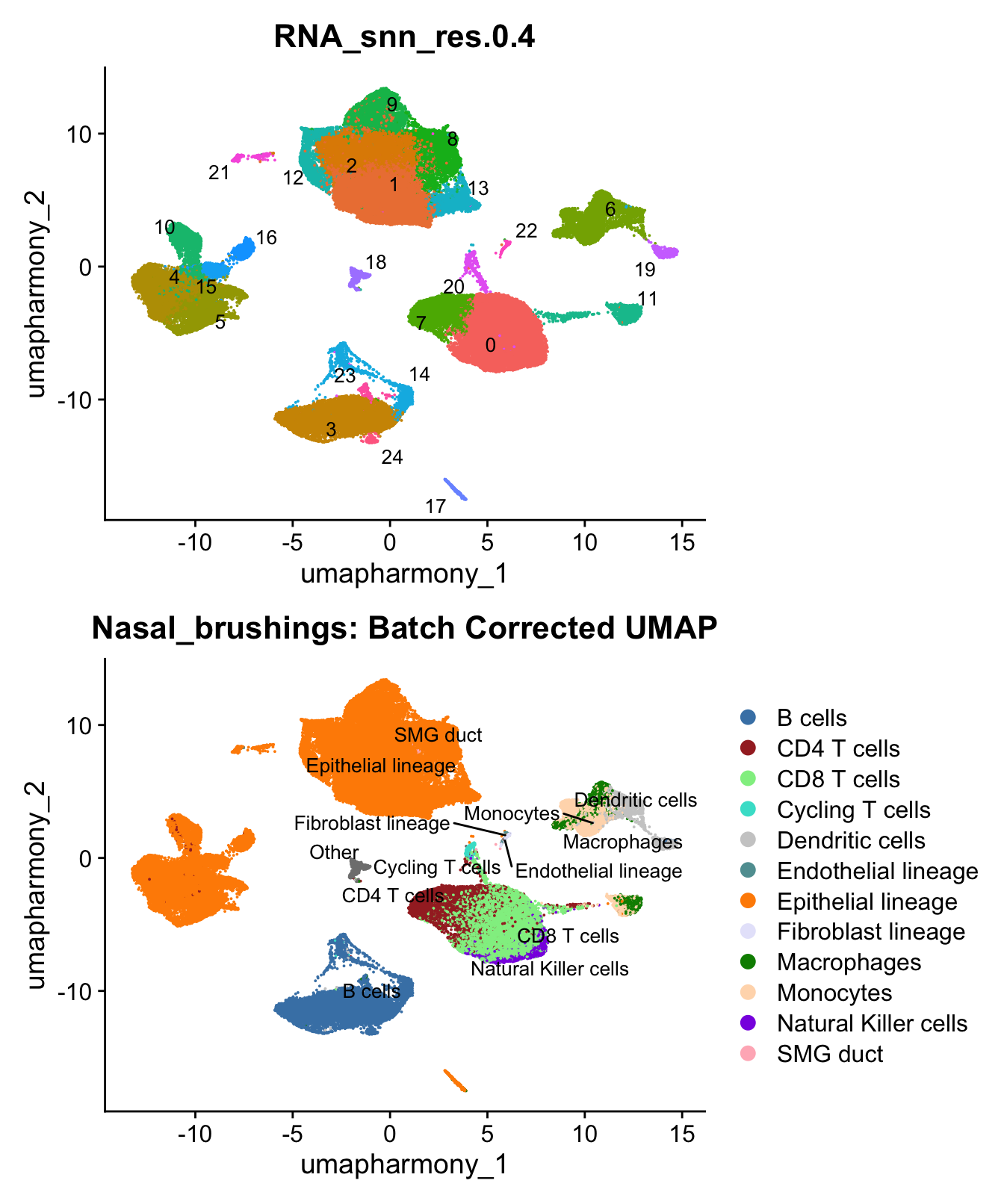
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Save batch corrected Object
out1 <- here("output",
"RDS", "AllBatches_Clustering_SEUs_v2",
paste0("G000231_Neeland_",tissue,".Clusters.SEU.rds"))
#dir.create(out1)
if (!file.exists(out1)) {
saveRDS(merged_obj, file = out1)
}Marker Gene Analysis
merged_obj <- JoinLayers(merged_obj)
paed.markers <- FindAllMarkers(merged_obj, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Extracting top 5 genes per cluster for visualization. The ‘top5’ contains the top 5 genes with the highest weighted average avg_log2FC within each cluster and the ‘best.wilcox.gene.per.cluster’ contains the single best gene with the highest weighted average avg_log2FC for each cluster.
paed.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "CCL5" "EPAS1" "ALPL" "CD79A" "ERICH3" "CCDC78" "SPI1" "TRAC"
[9] "CXCL10" "FOSB" "CDC20B" "CSF3R" "SPRR3" "TK1" "CD79A" "FRMPD2"
[17] "FRMPD2" "HBB" "CPA3" "LILRA4" "IL2RB" "FOXI1" "MLANA" "G0S2"
[25] "MZB1" Marker gene expression in clusters
This heatmap depicts the expression of top five genes in each cluster.
DoHeatmap(merged_obj, features = top5$gene) + NoLegend()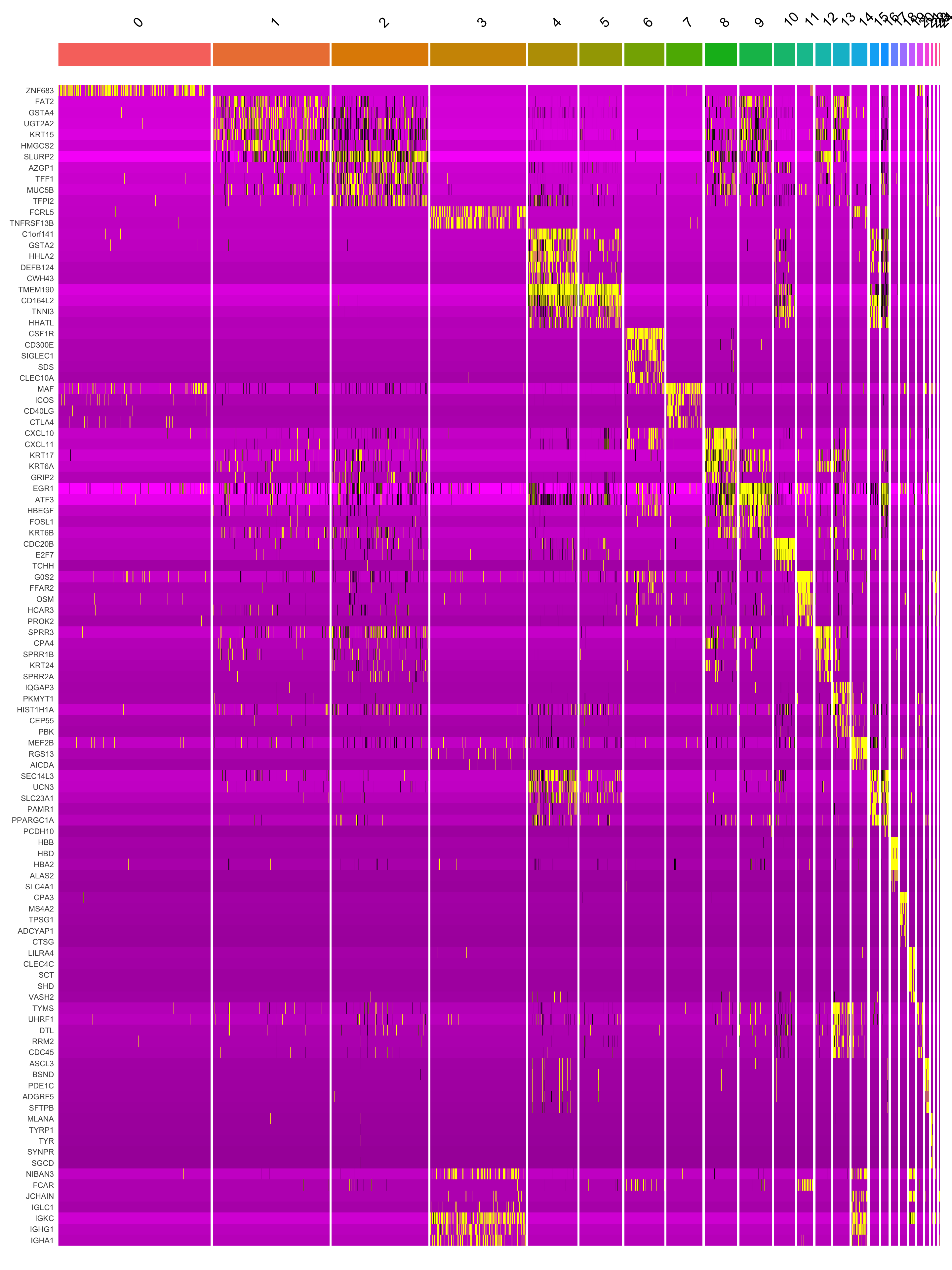
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Violin plot shows the expression of top marker gene per cluster.
VlnPlot(merged_obj, features=best.wilcox.gene.per.cluster, ncol = 2, raster = FALSE, pt.size = FALSE)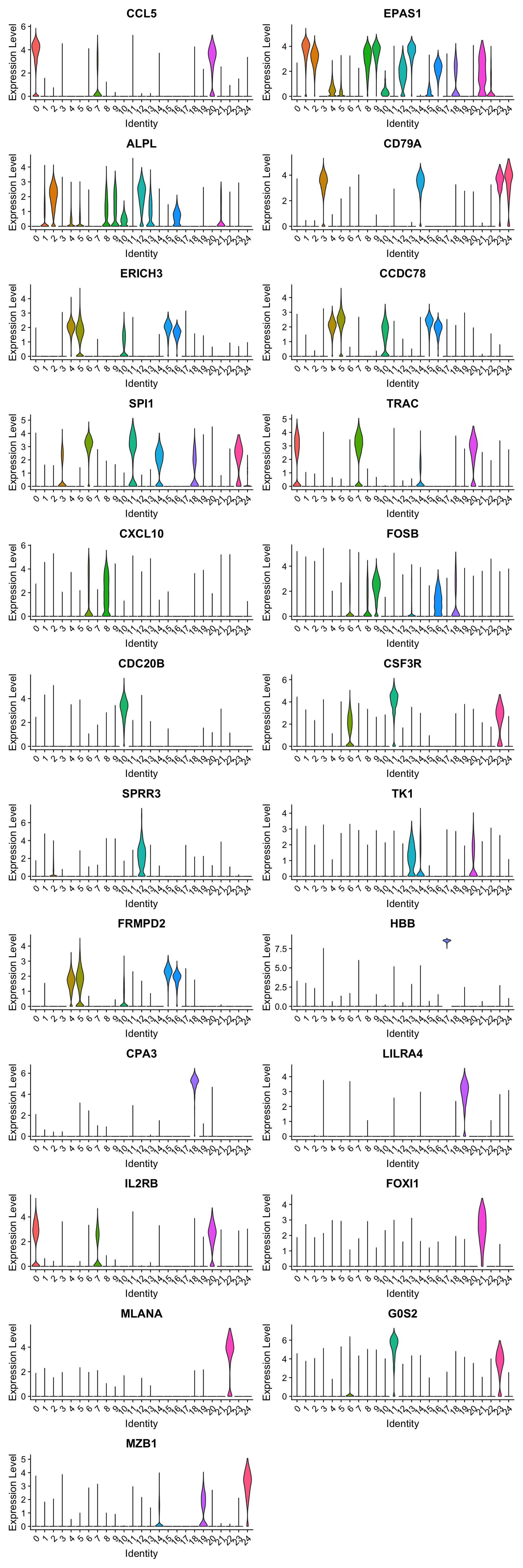
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Violin plot shows the expression of top marker gene per cluster and compares its expression in both batches.
plots <- VlnPlot(merged_obj, features = best.wilcox.gene.per.cluster, split.by = "batch_name", group.by = "Broad_cell_label_3",
pt.size = 0, combine = FALSE, raster = FALSE, split.plot = TRUE)The default behaviour of split.by has changed.
Separate violin plots are now plotted side-by-side.
To restore the old behaviour of a single split violin,
set split.plot = TRUE.
This message will be shown once per session.wrap_plots(plots = plots, ncol = 1)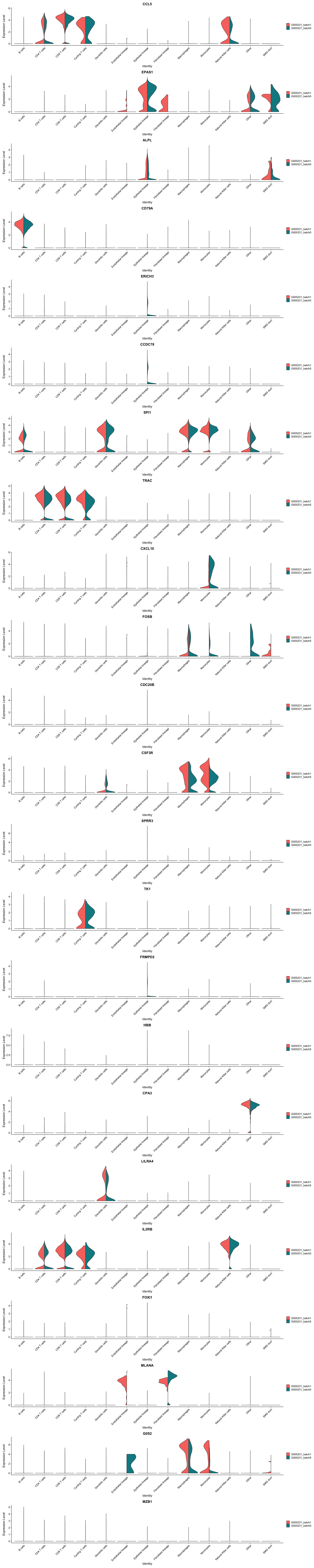
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(merged_obj,features=best.wilcox.gene.per.cluster, reduction = 'umap.harmony', raster = FALSE, ncol = 2)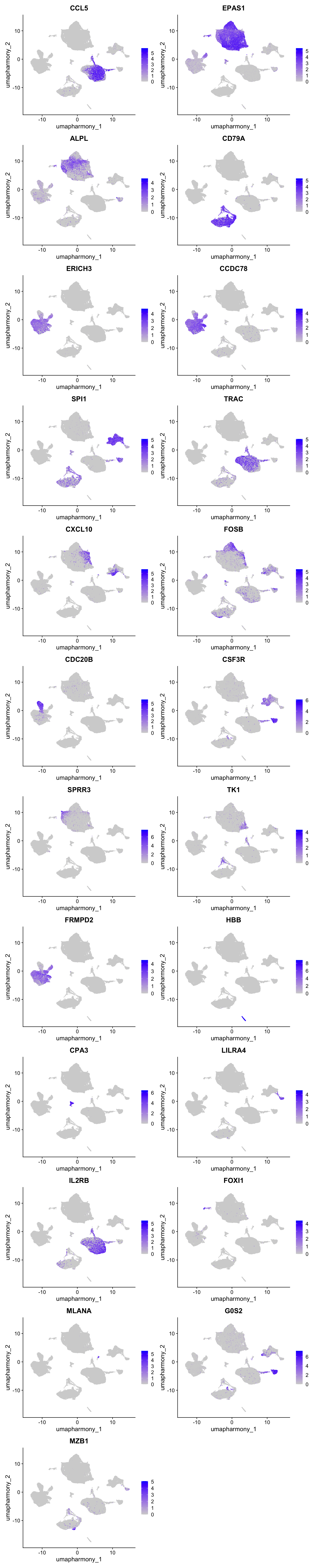
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Extract markers for each cluster
This section extracts marker genes for each cluster and save them as a CSV file.
out_markers <- here("output",
"CSV_v2", tissue,
paste(tissue,"_Marker_gene_clusters.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed.markers$cluster)) {
cluster_data <- paed.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
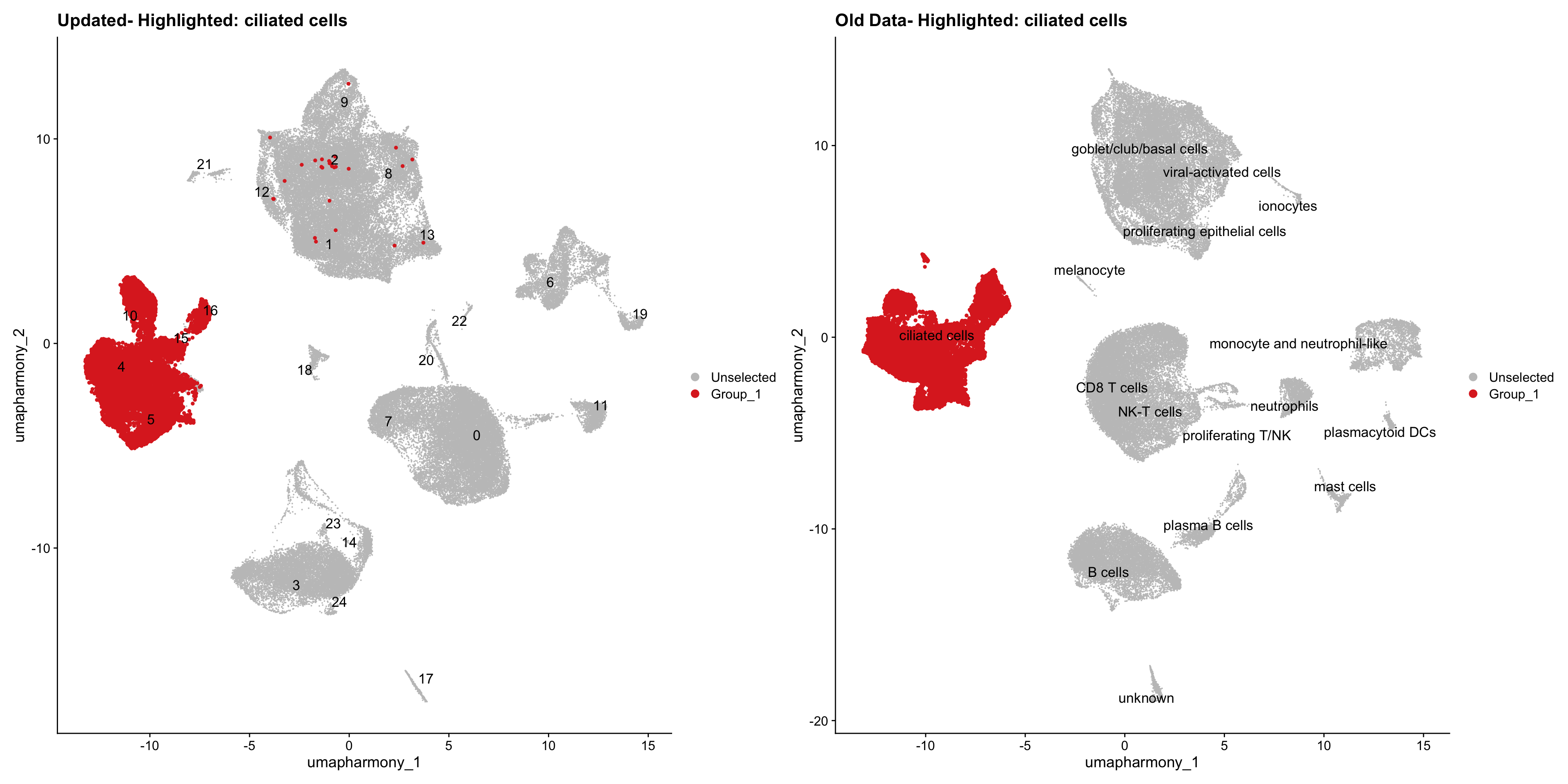
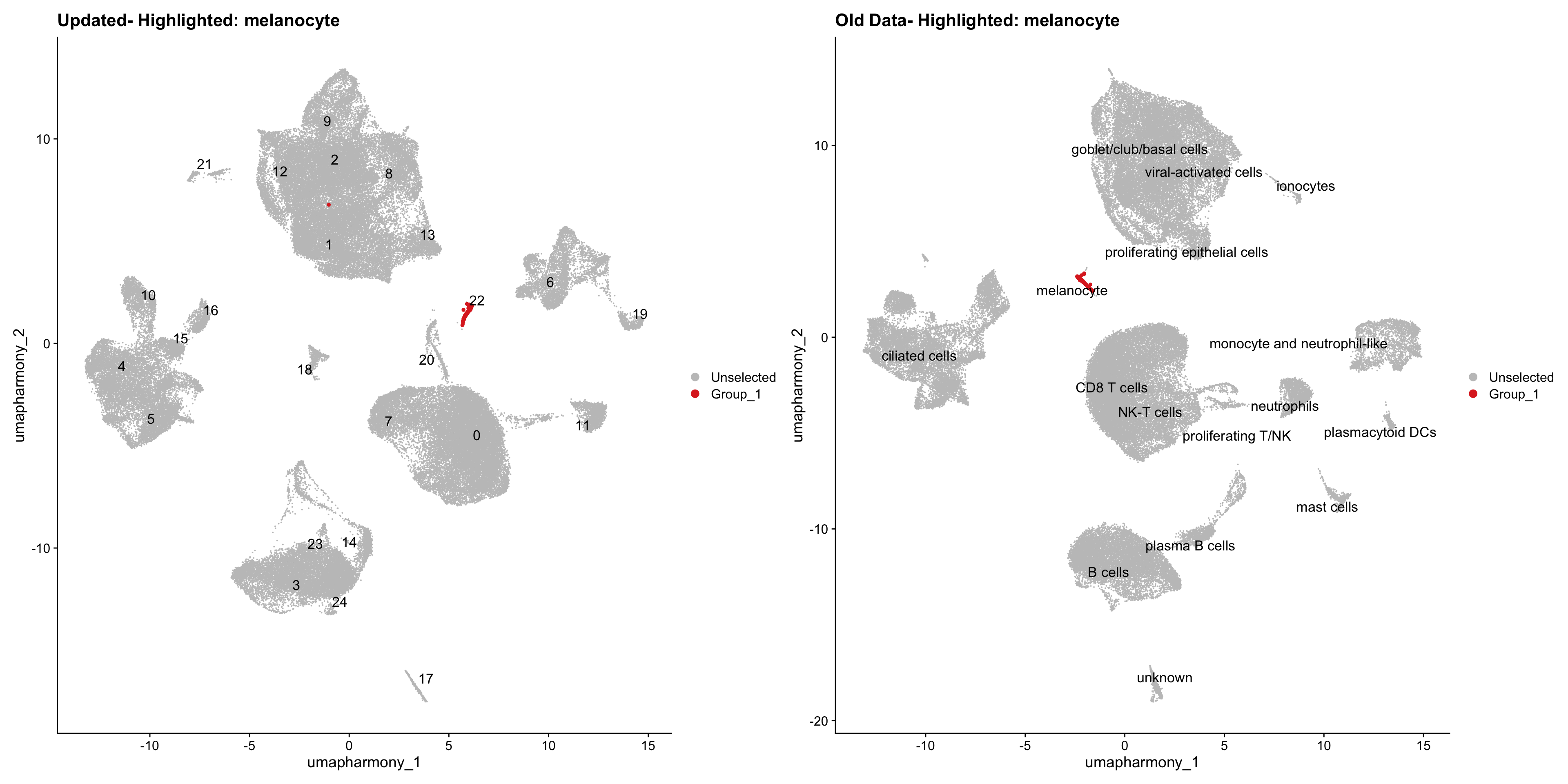
}Using old labels to annotate cell types
out1 <- here("output",
"RDS", "AllBatches_Clustering_SEUs",
paste0("G000231_Neeland_",tissue,".Clusters.SEU.rds"))
old_obj <- readRDS(out1)cell_types <- unique(old_obj$cell_labels)
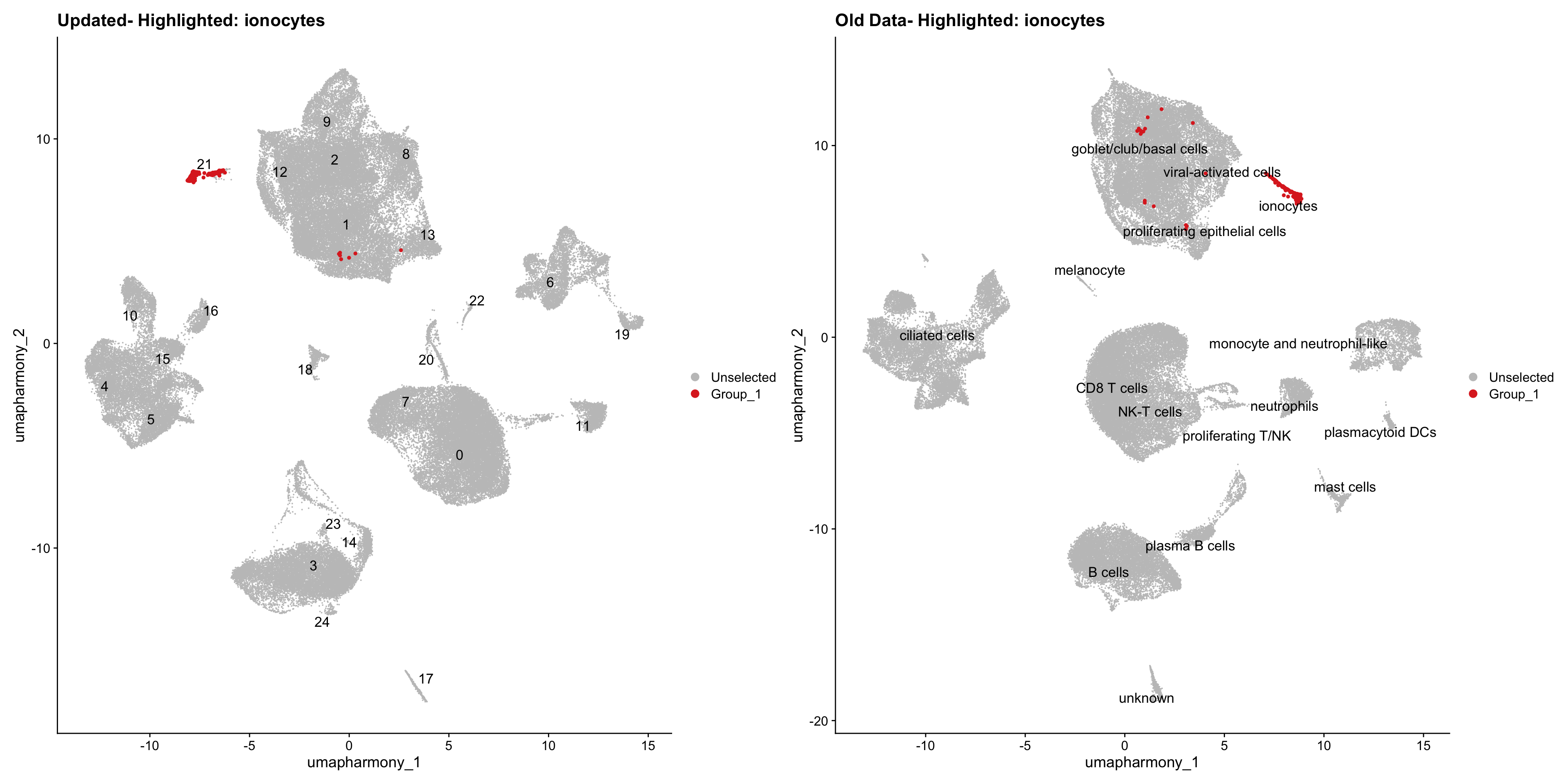
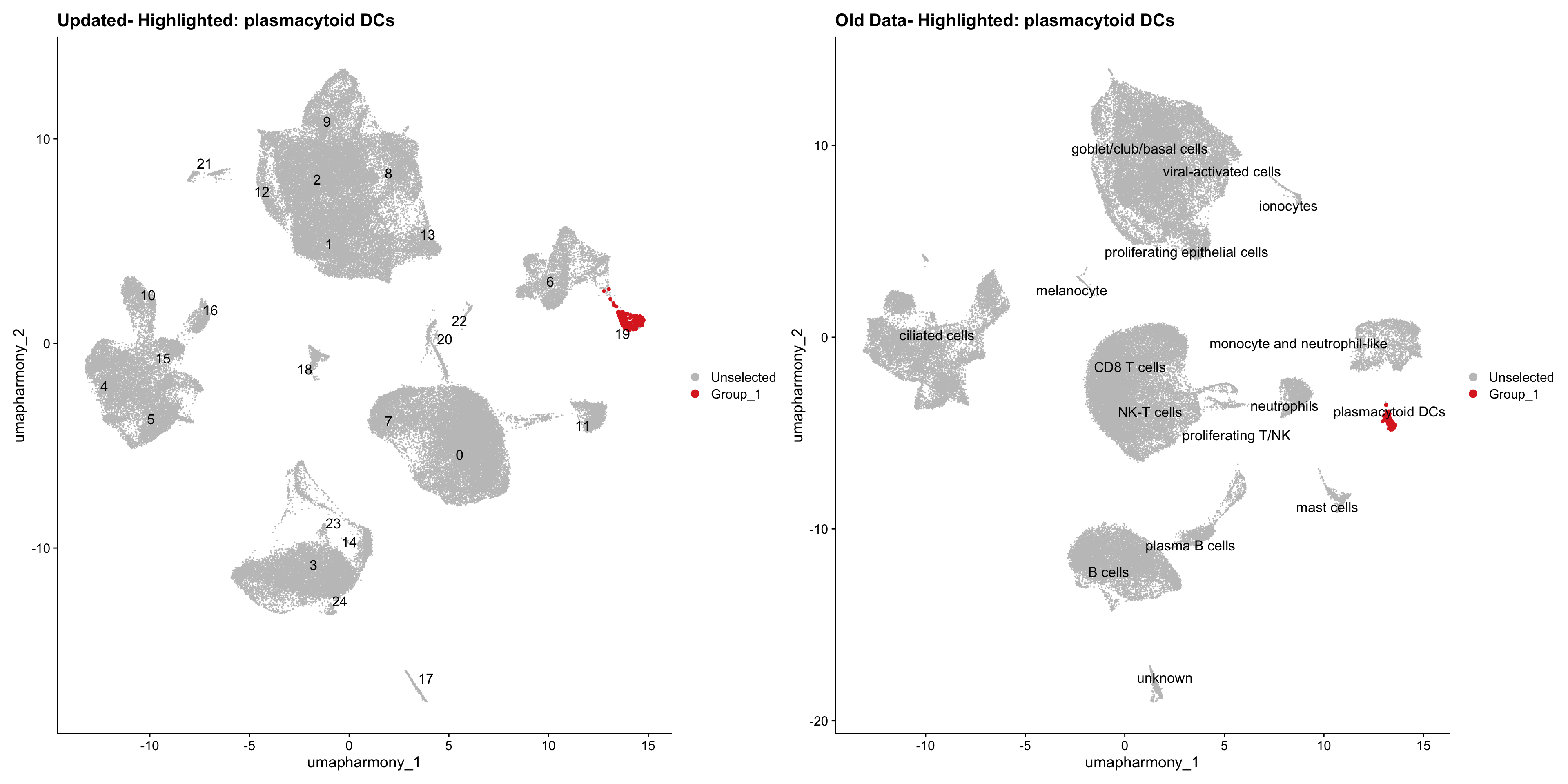
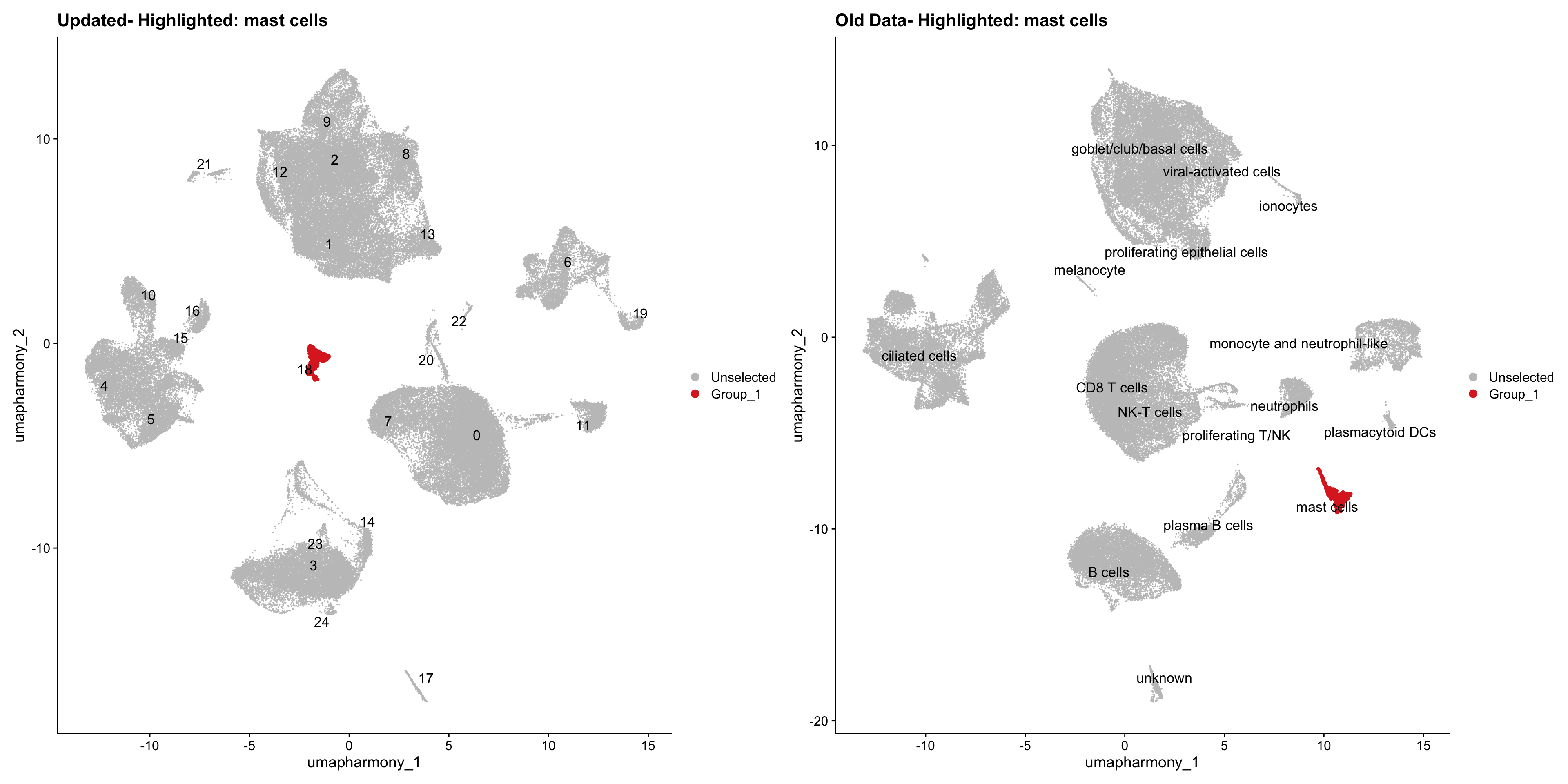
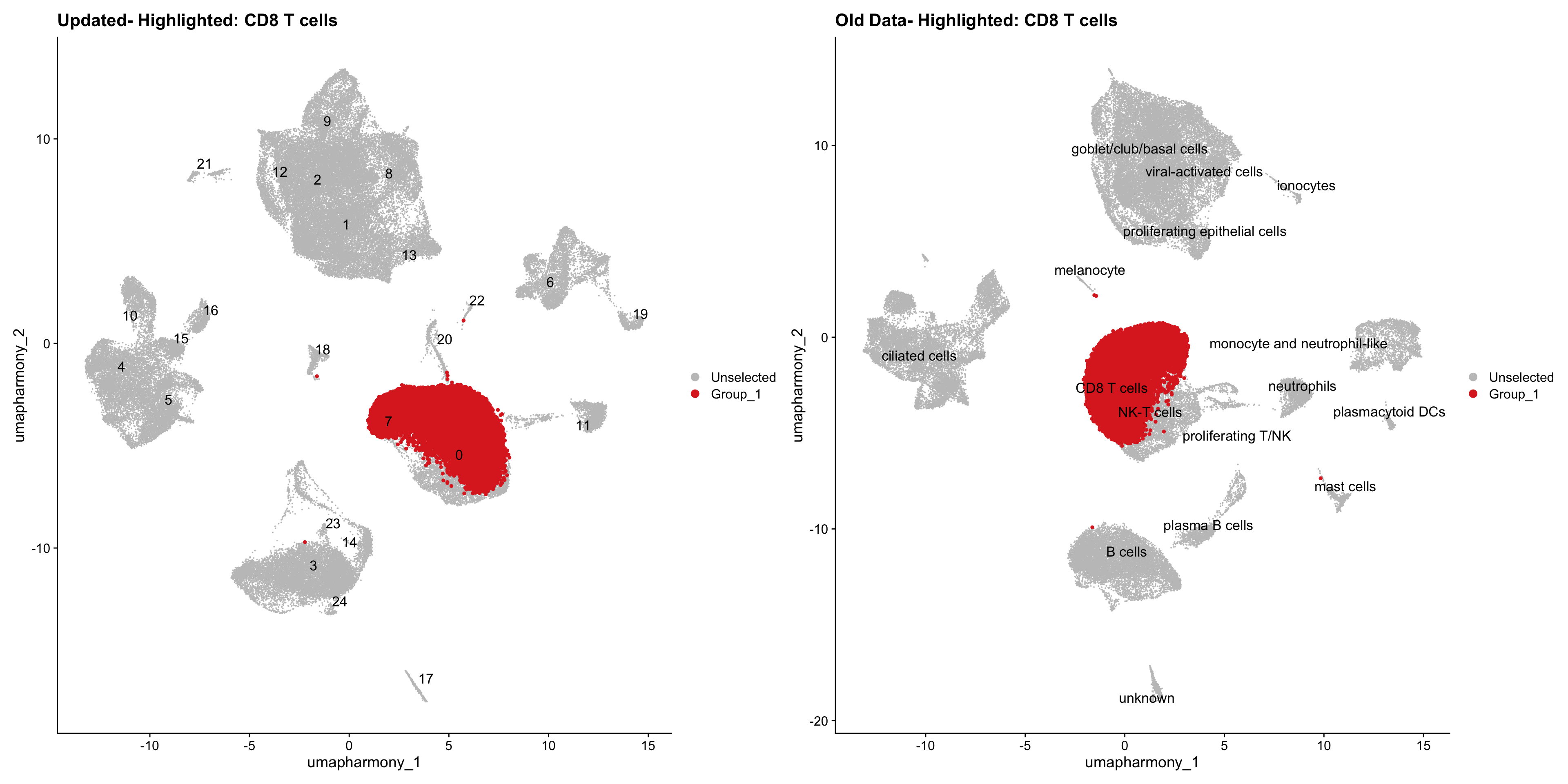
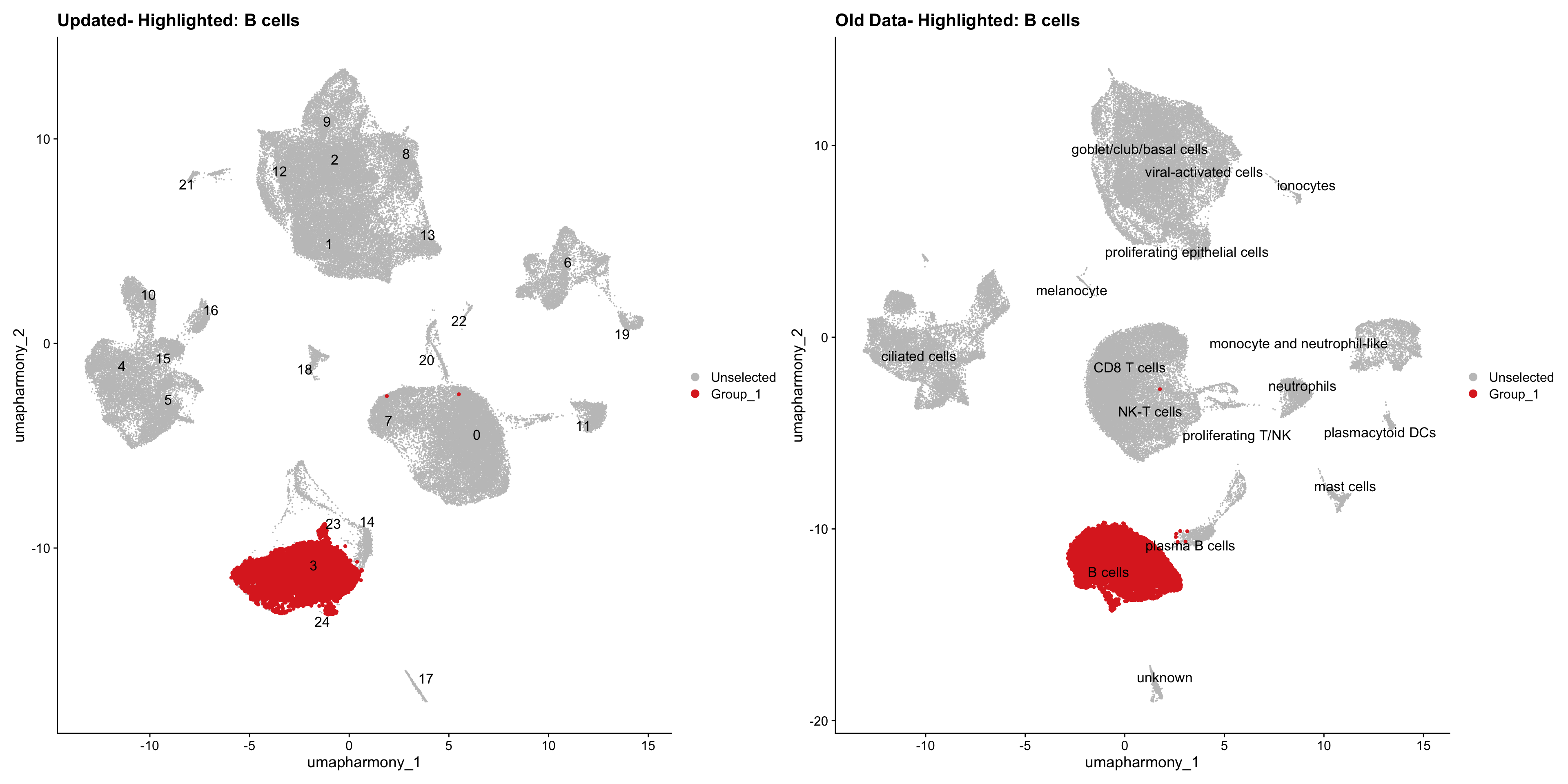
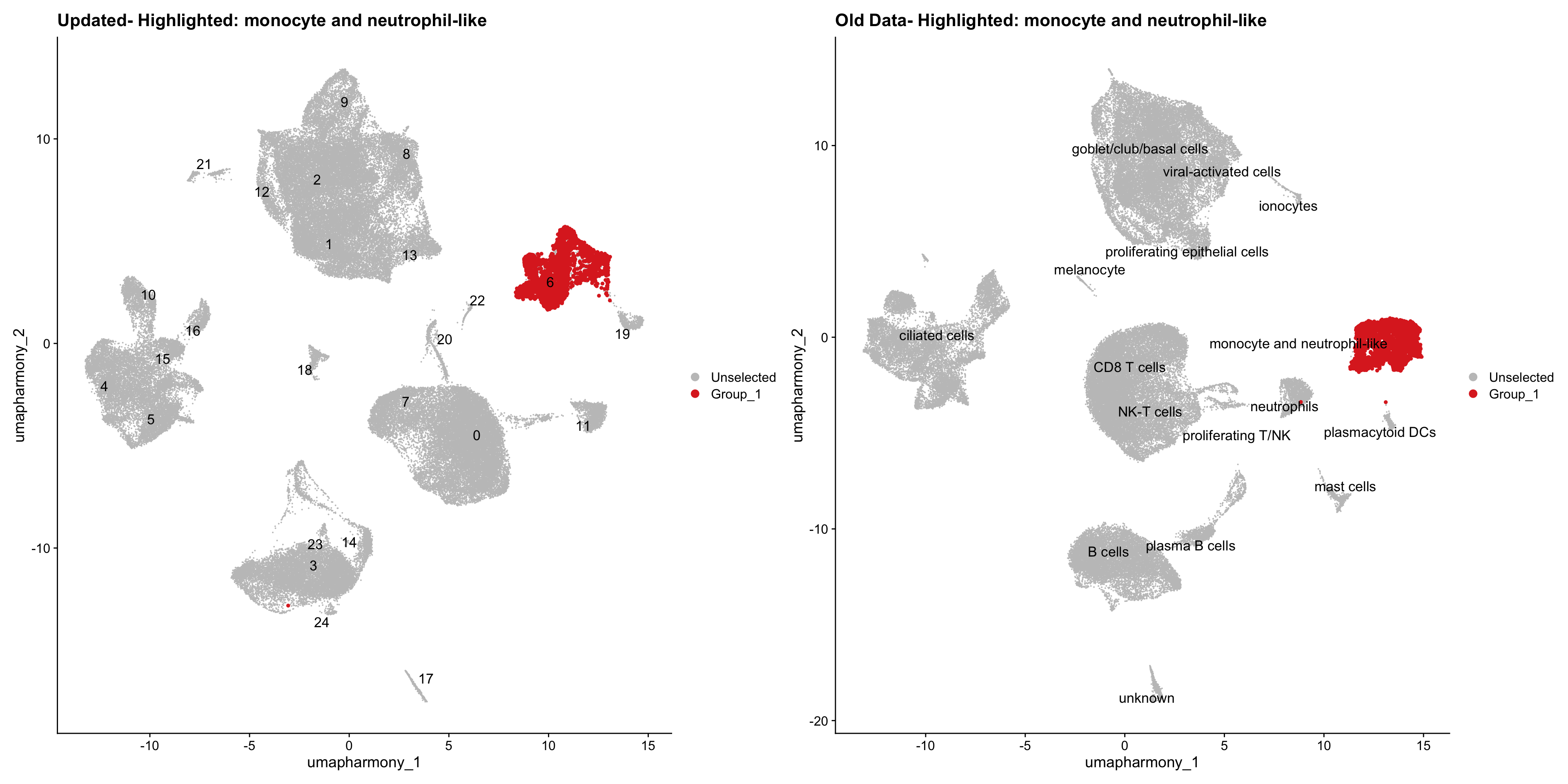
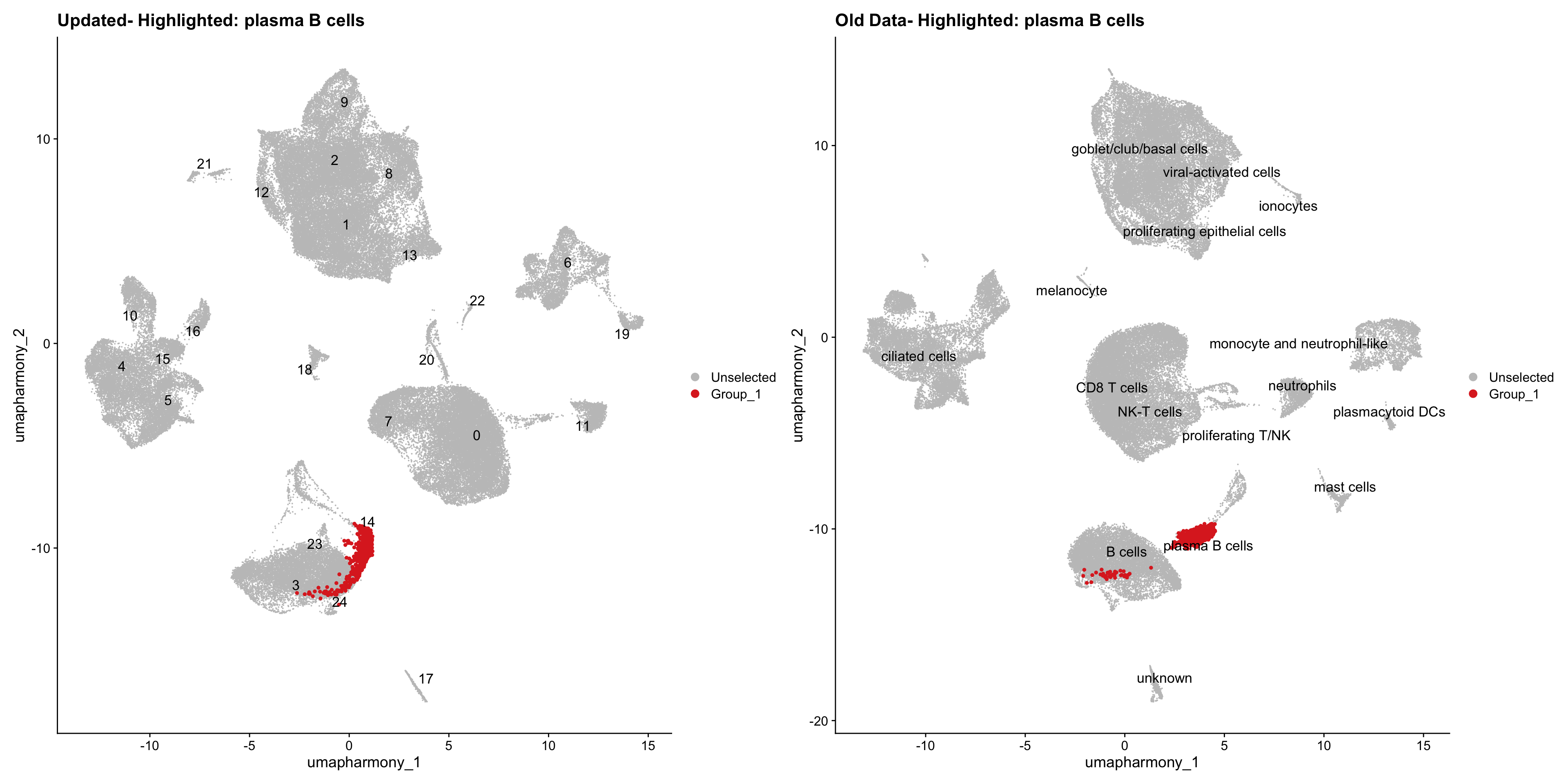
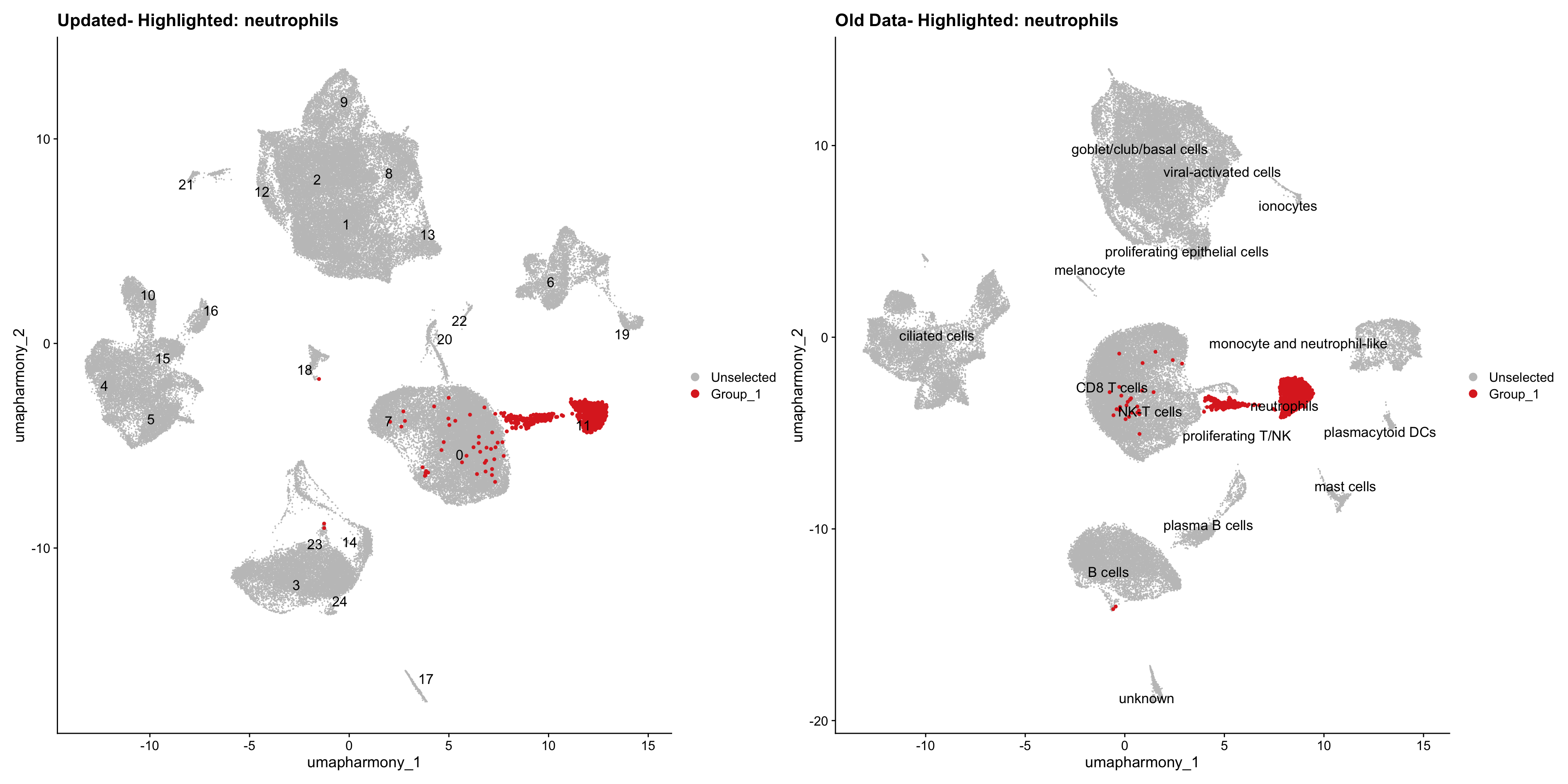
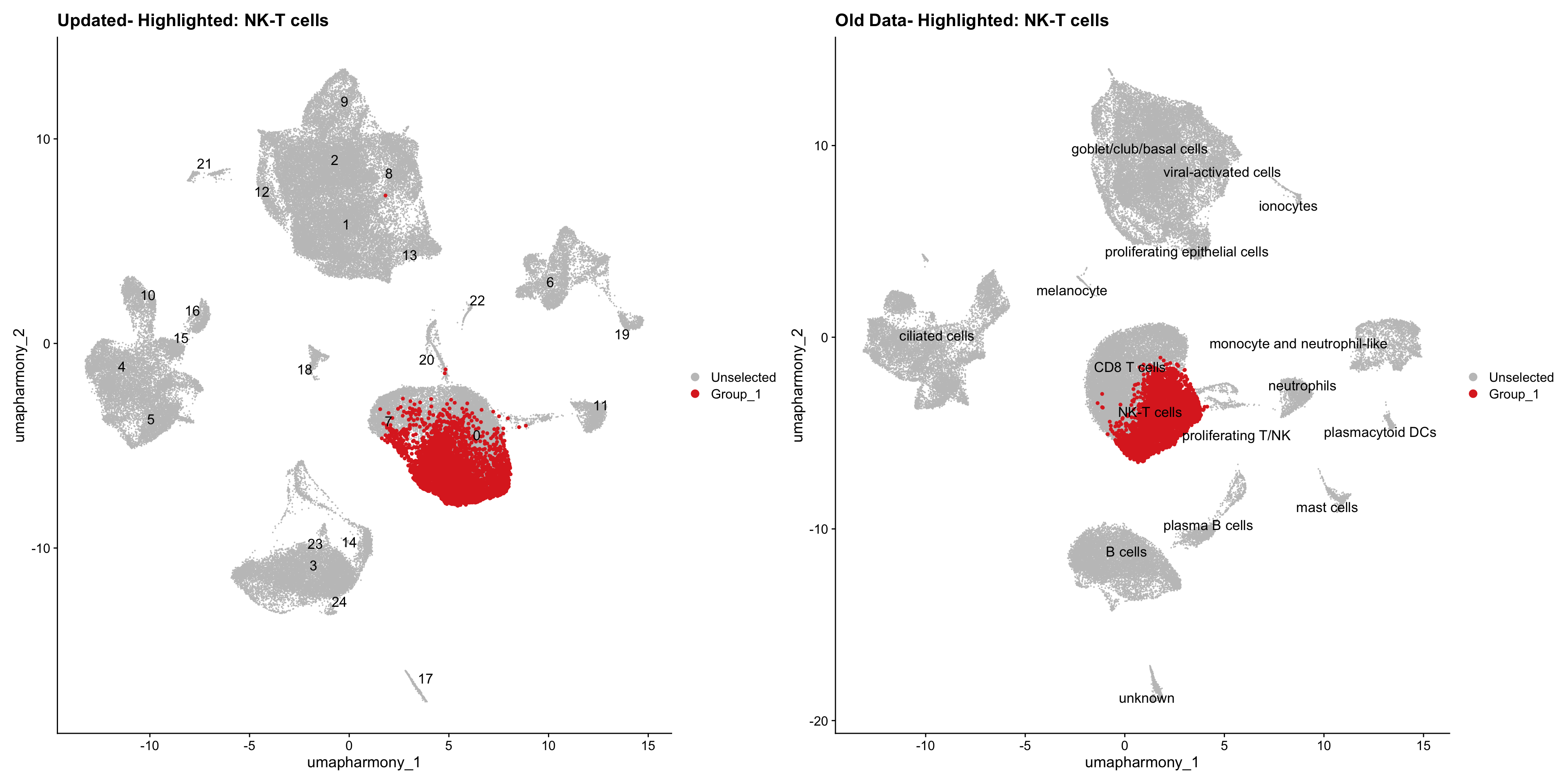
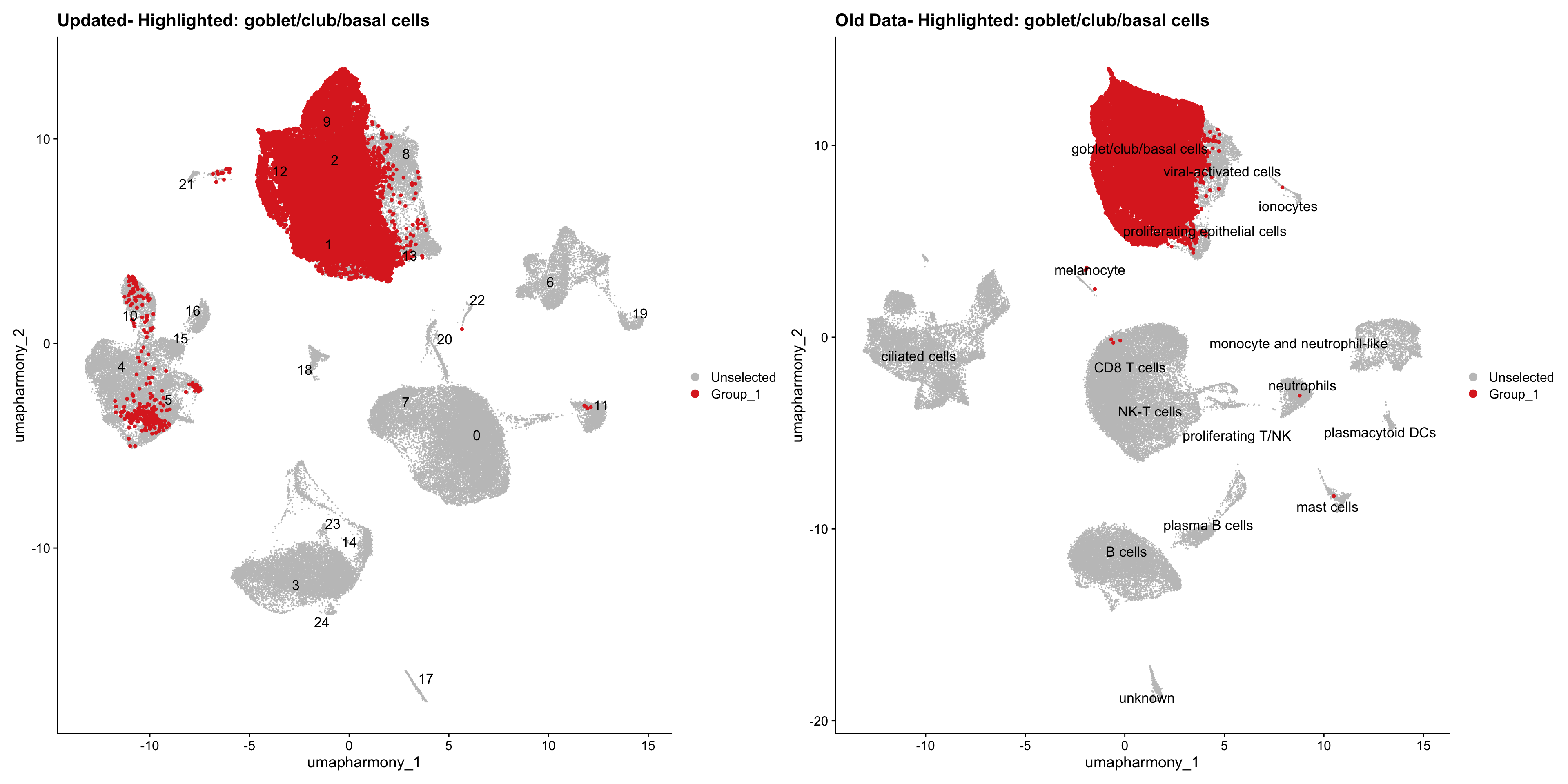
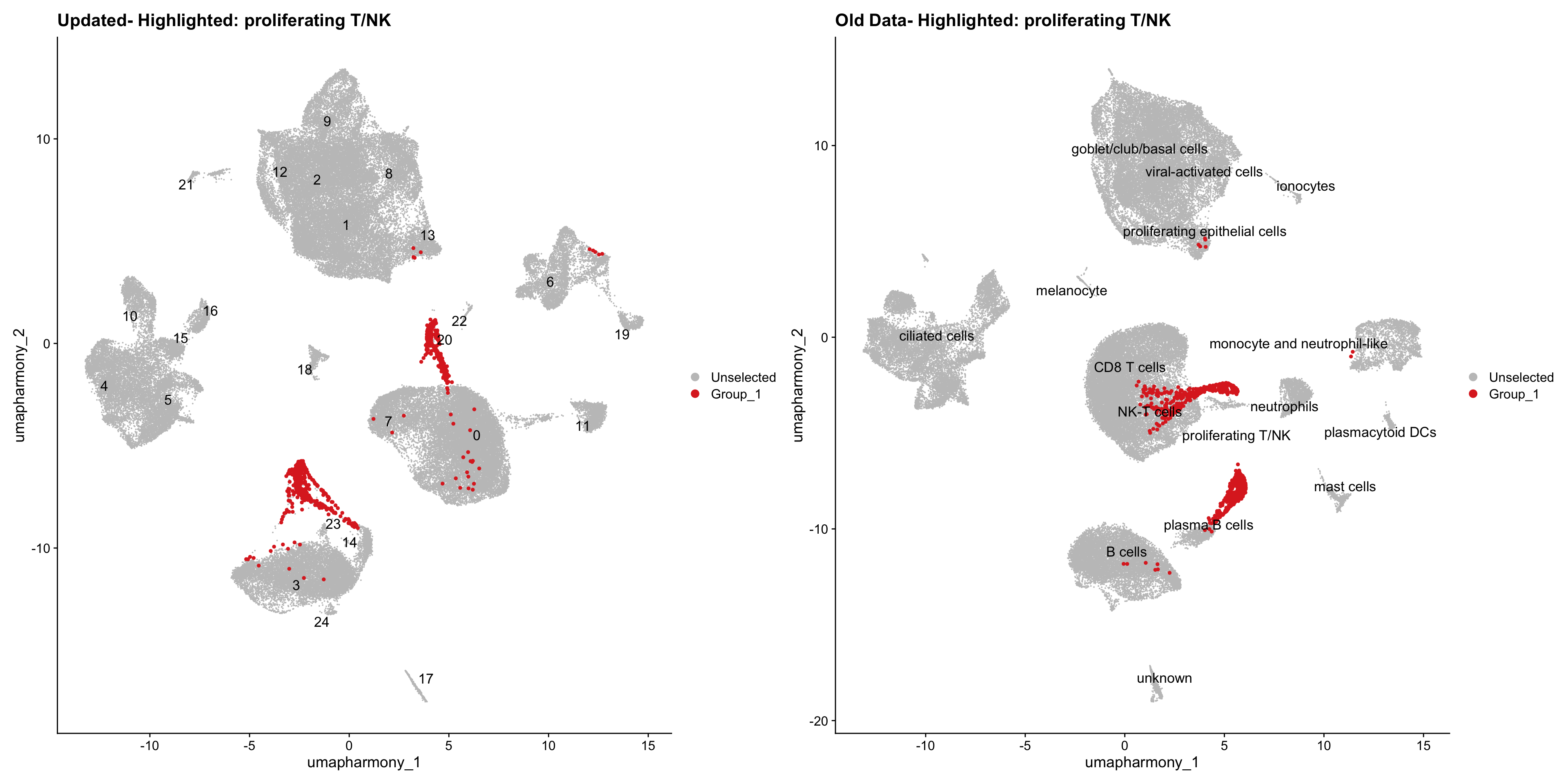
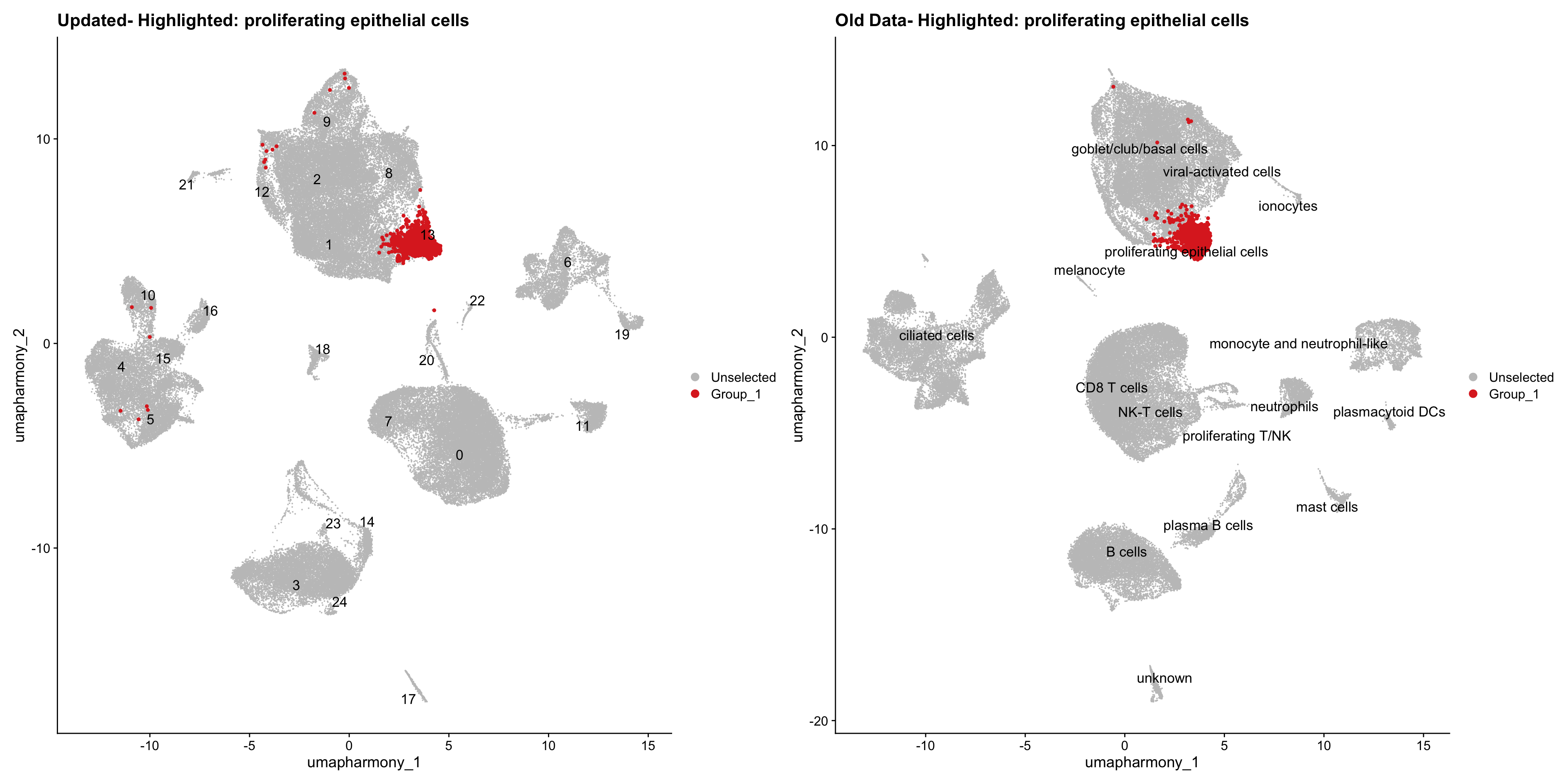
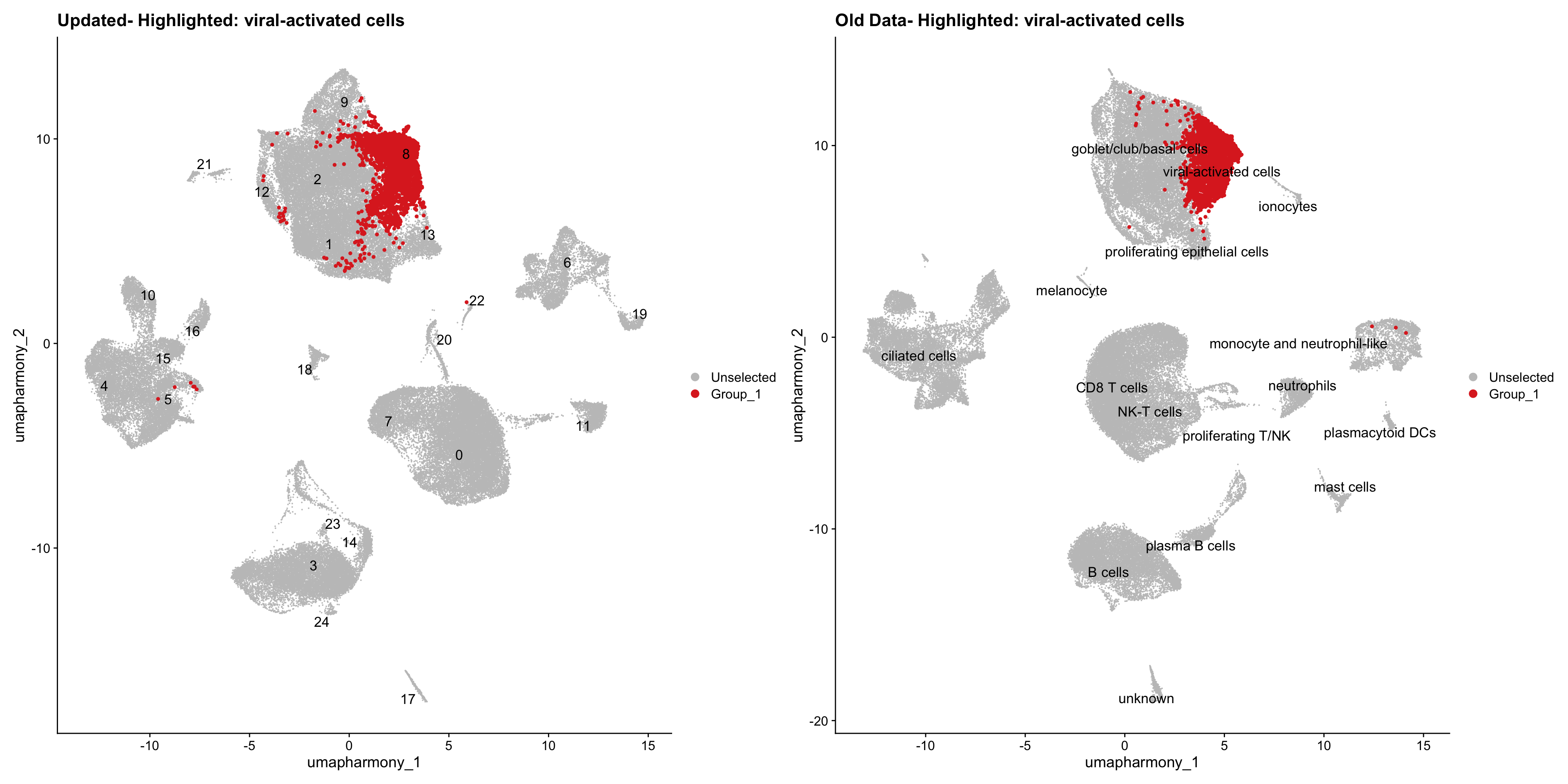
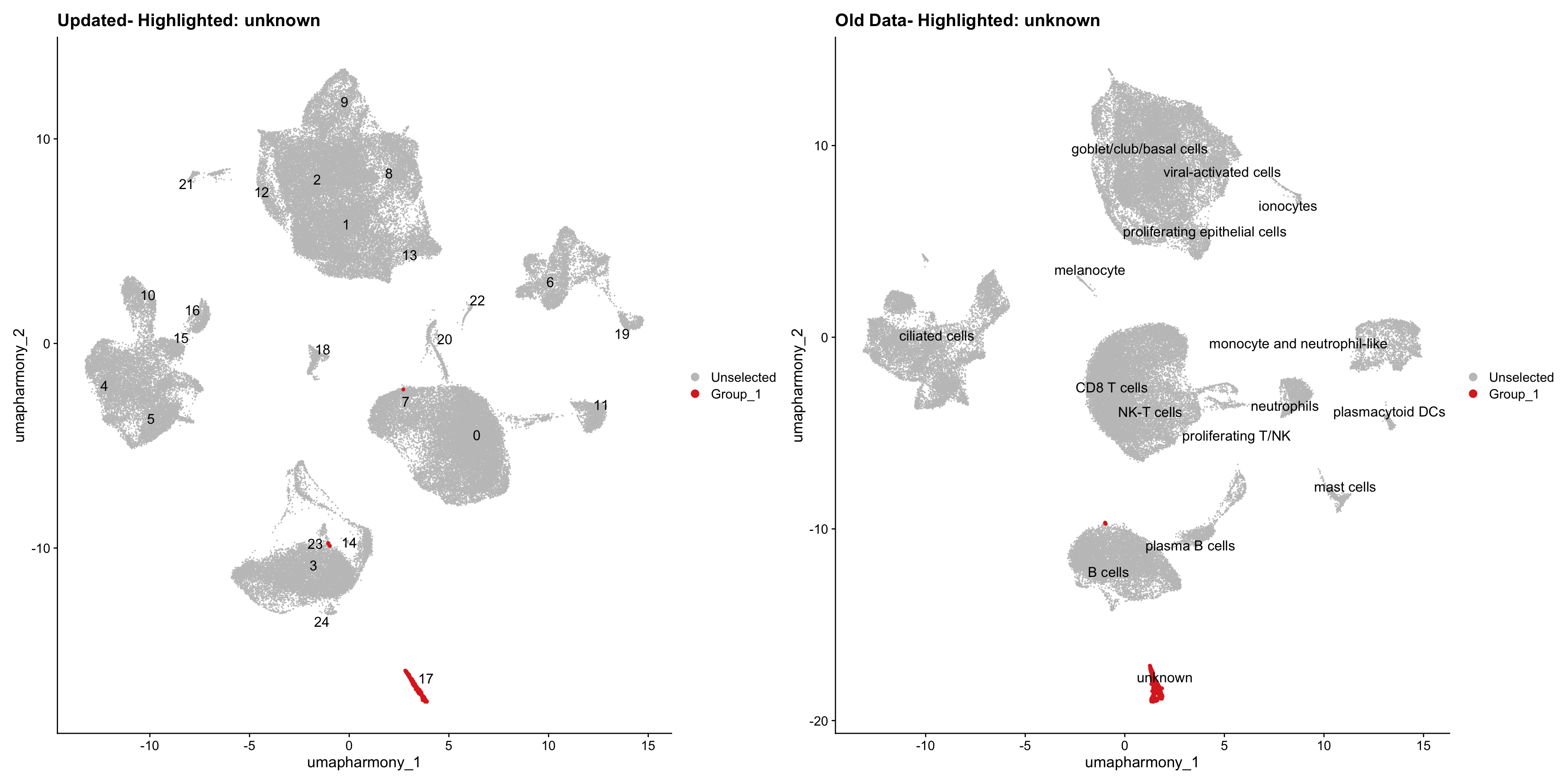
for (cell_type in cell_types) {
cl_cells <- WhichCells(old_obj, idents = cell_type)
p <- DimPlot(
merged_obj,
reduction = "umap.harmony",
label = TRUE,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Updated- Highlighted:", cell_type))
p1 <- DimPlot(
old_obj,
reduction = "umap.harmony",
label = T,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Old Data- Highlighted:", cell_type))
print(p | p1)
}
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
| Version | Author | Date |
|---|---|---|
| eebc9b9 | Gunjan Dixit | 2024-12-22 |
Updated cell-type labels (all clusters)
cell_labels <- readxl::read_excel(here("data/cell_labels_Mel_v4_Dec2024/earlyAIR_NB_all.xlsx"), sheet = "all_clusters")
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
merged_obj <- RenameIdents(merged_obj, new_cluster_names)
merged_obj@meta.data$cell_labels <- Idents(merged_obj)
p3 <- DimPlot(merged_obj, reduction = "umap.harmony", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated cell types"))
p3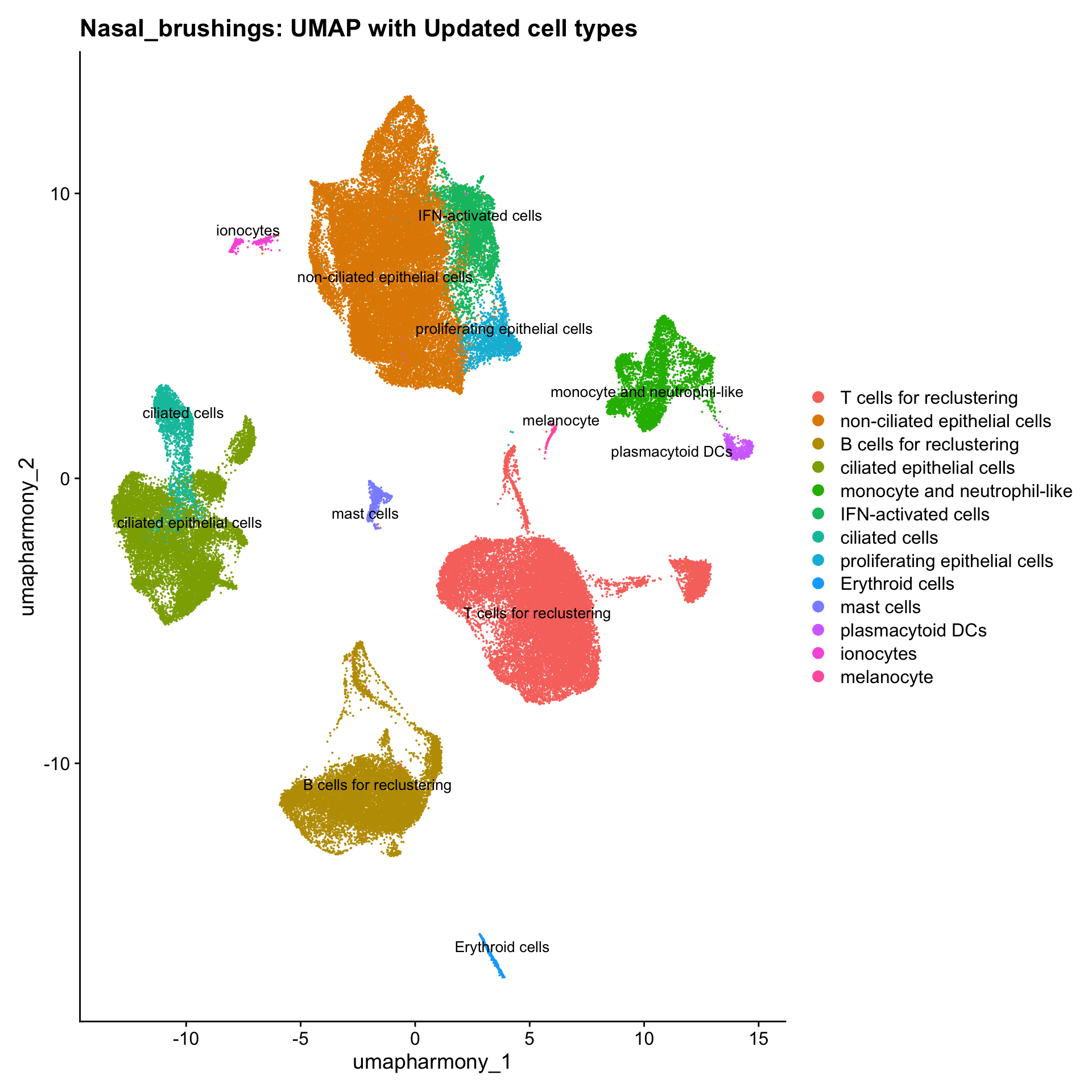
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
Reclustering T cell population
The marker genes for this reclustering can be found here-
#sub_clusters <- c(0,7,11,20)
#idx <- which(merged_obj$cluster %in% sub_clusters)
idx <- which(Idents(merged_obj) %in% "T cells for reclustering")
paed_sub <- merged_obj[,idx]
mito_genes <- grep("^MT-", rownames(paed_sub), value = TRUE)
paed_sub <- subset(paed_sub, features = setdiff(rownames(paed_sub), mito_genes))
paed_subAn object of class Seurat
17962 features across 23715 samples within 1 assay
Active assay: RNA (17962 features, 2000 variable features)
3 layers present: data, counts, scale.data
4 dimensional reductions calculated: pca, umap.unintegrated, harmony, umap.harmonypaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.tcell")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9511
Number of communities: 7
Elapsed time: 16 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9269
Number of communities: 9
Elapsed time: 13 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9139
Number of communities: 12
Elapsed time: 13 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9026
Number of communities: 13
Elapsed time: 13 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8922
Number of communities: 15
Elapsed time: 12 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8829
Number of communities: 17
Elapsed time: 12 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8744
Number of communities: 17
Elapsed time: 12 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8663
Number of communities: 19
Elapsed time: 12 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8587
Number of communities: 20
Elapsed time: 12 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 23715
Number of edges: 779864
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8514
Number of communities: 21
Elapsed time: 11 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)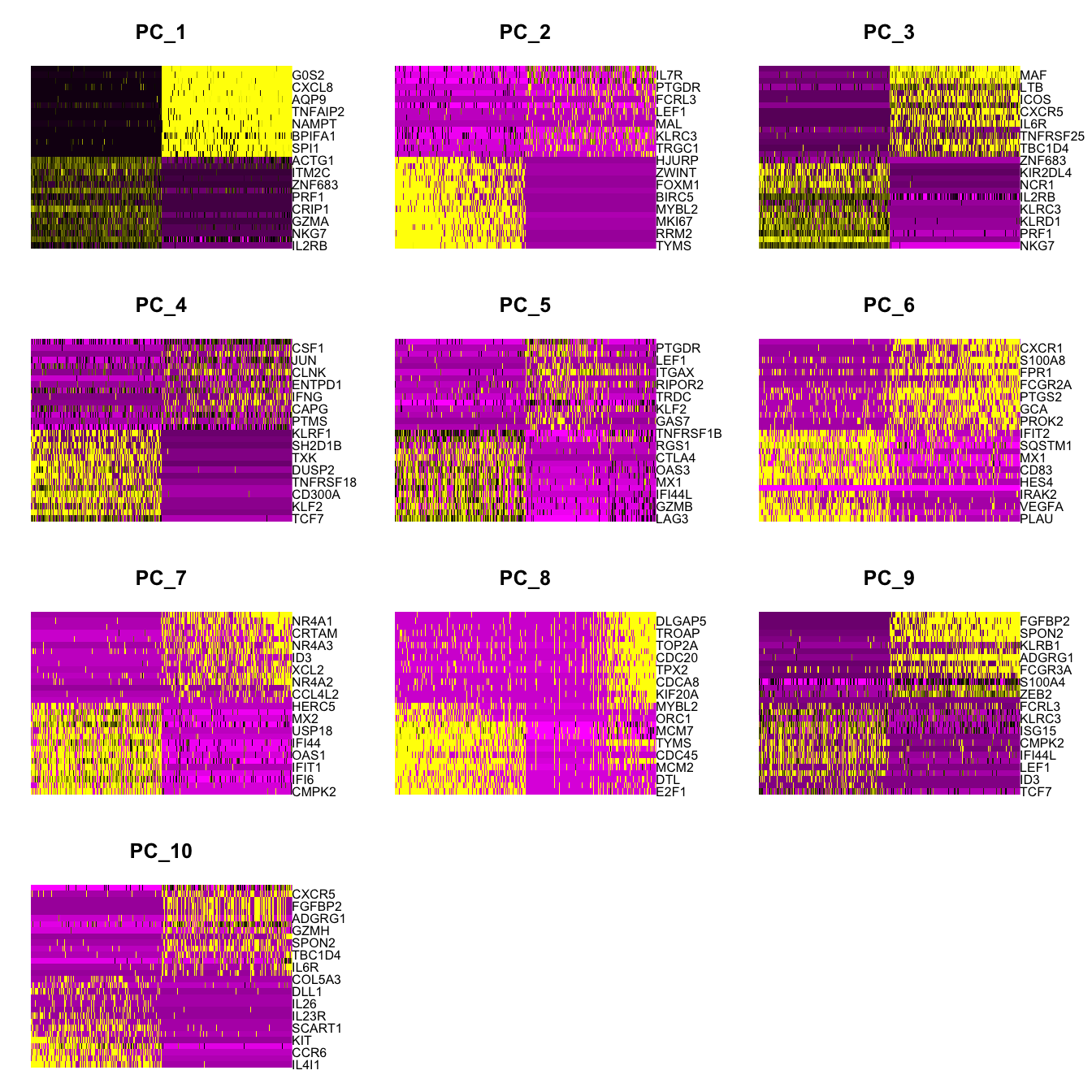
clustree(paed_sub, prefix = "RNA_snn_res.")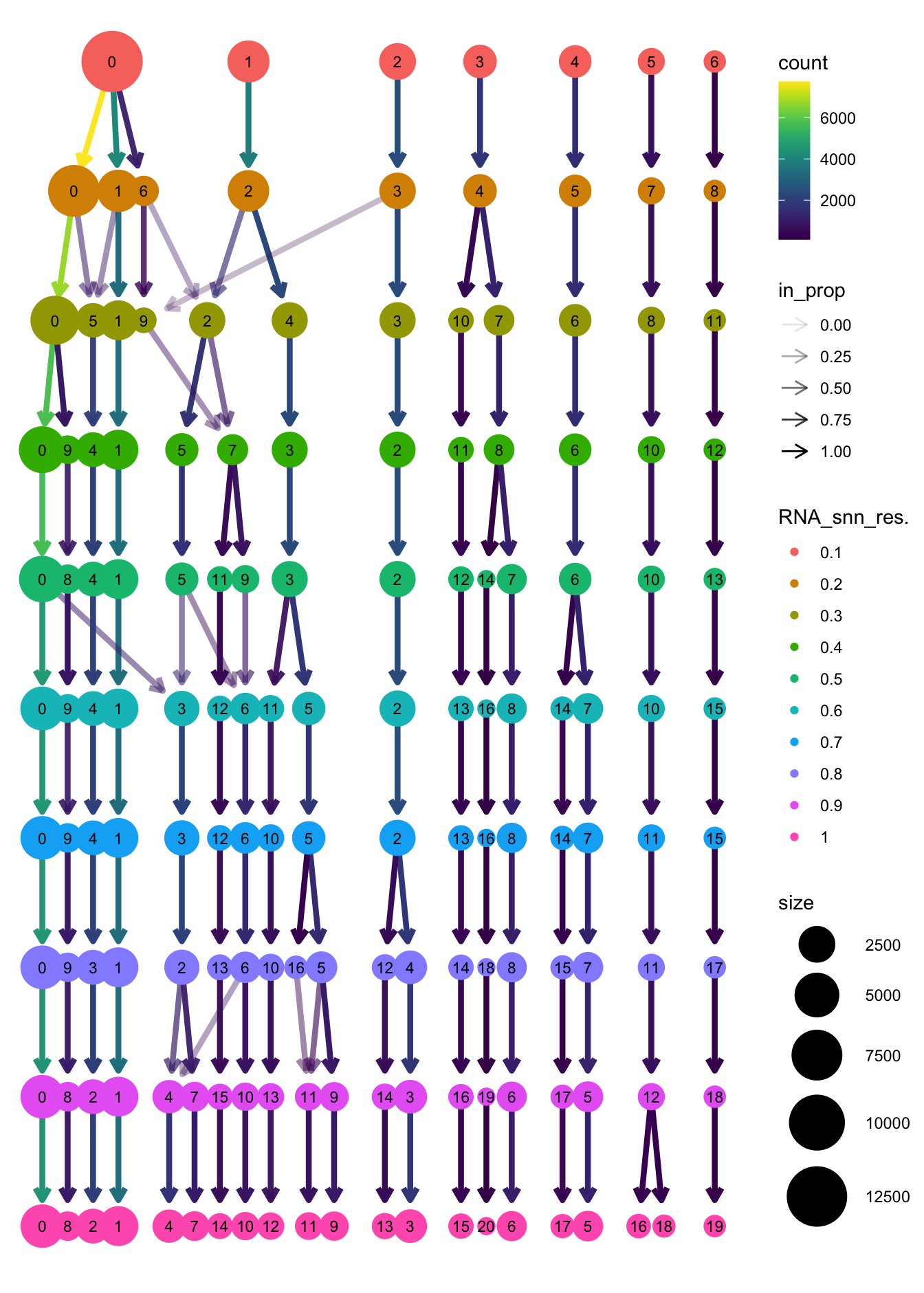
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(paed_sub$RNA_snn_res.0.4)
paed_sub$RNA_snn_res.0.4 <- factor(paed_sub$RNA_snn_res.0.4, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
Idents(paed_sub) <- paed_sub$RNA_snn_res.0.4DimPlot(paed_sub, reduction = "umap.tcell", group.by = "RNA_snn_res.0.4", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )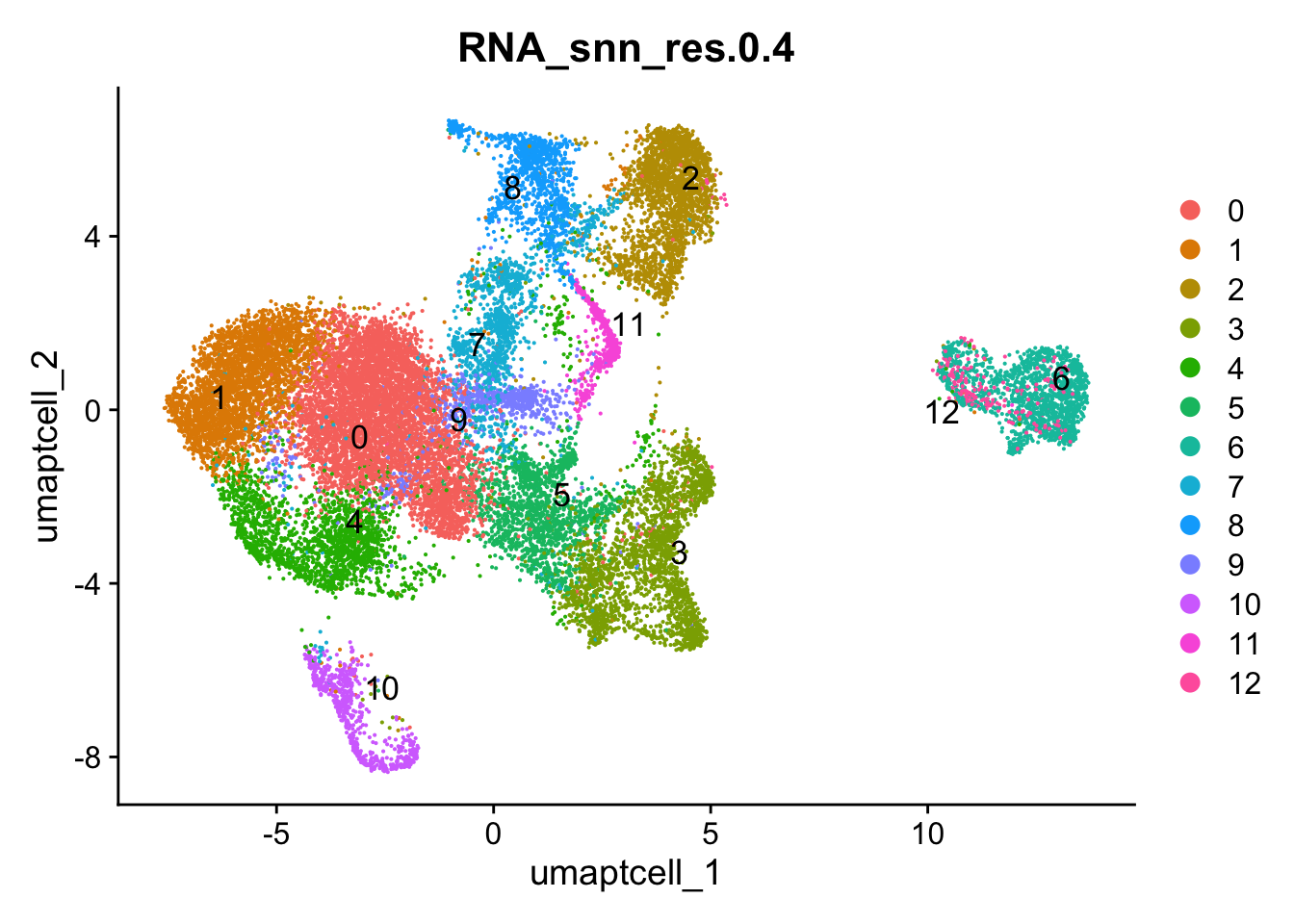
paed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8Calculating cluster 9Calculating cluster 10Calculating cluster 11Calculating cluster 12paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "CD8A" "CSF1" "TCF7" "CD4" "IFI6" "MAF" "CSF3R" "NR4A2" "GNLY"
[10] "GZMK" "TYMS" "KLF2" "CSF3R"FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.tcell', raster = FALSE, label = T, ncol = 3)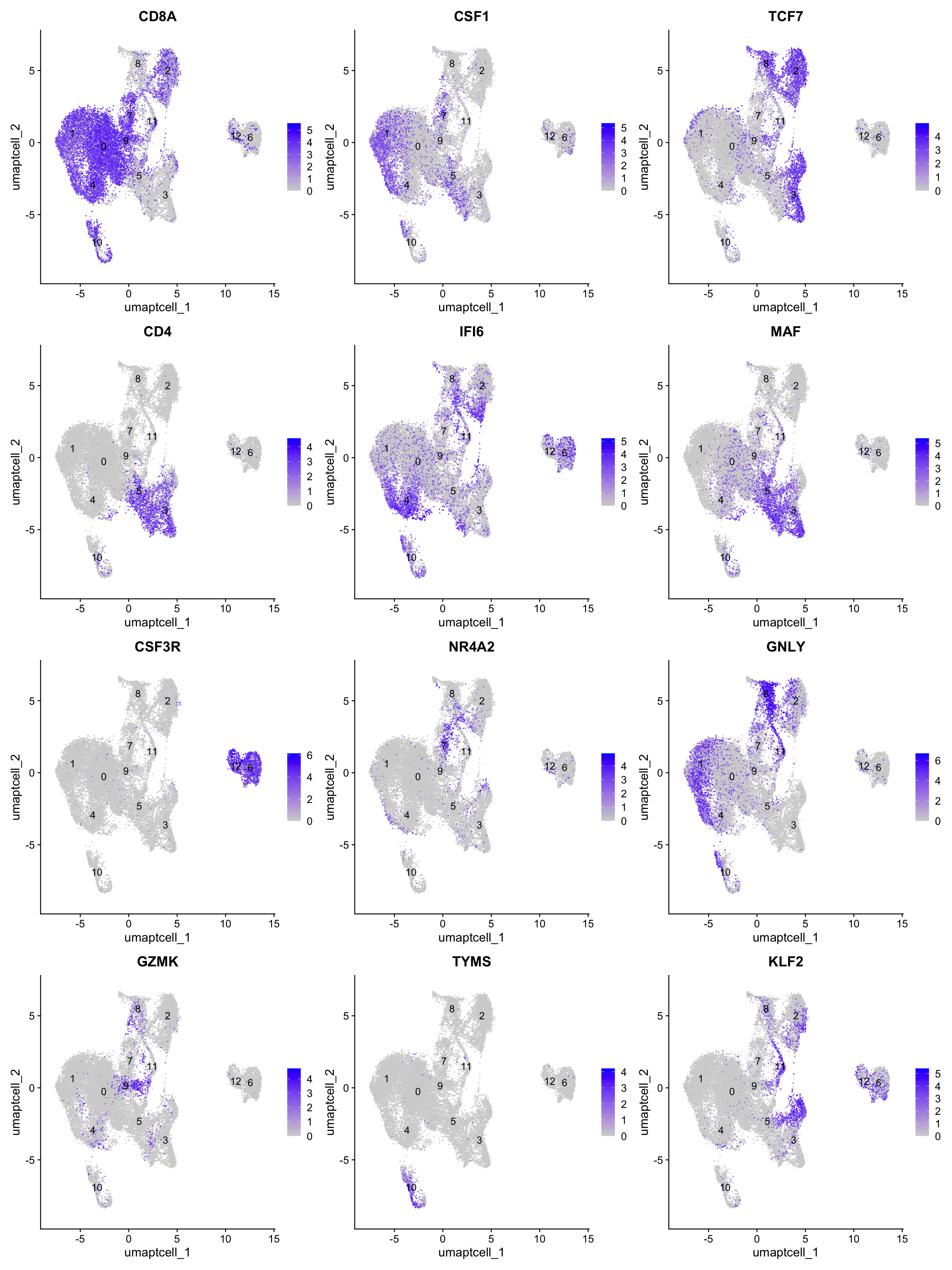
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.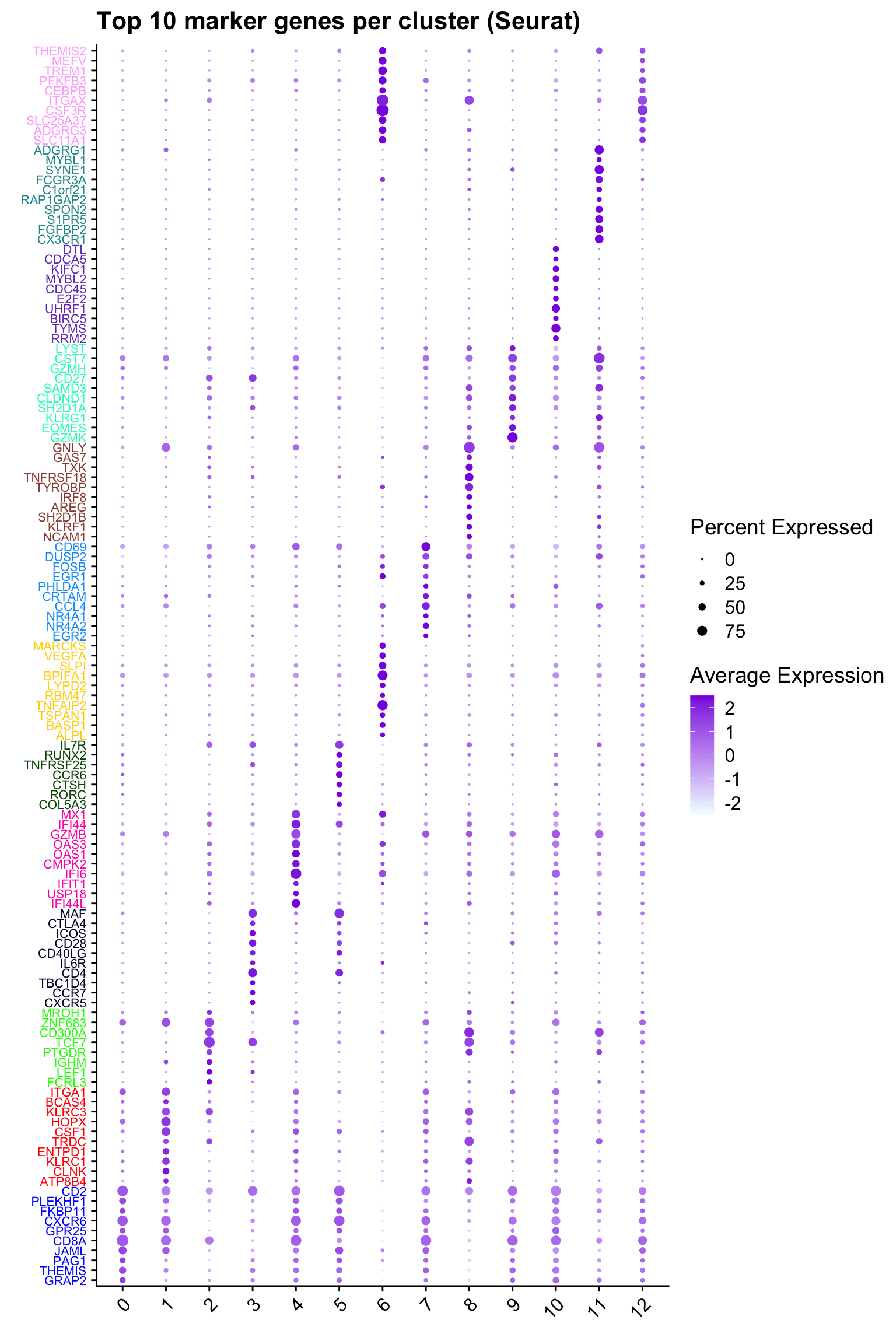
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
out_markers <- here("output",
"CSV_v2",tissue,
paste(tissue,"_Marker_genes_Reclustered_Tcell_population.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
if (!file.exists(file_name)) {
write.csv(cluster_data, file = file_name)
}
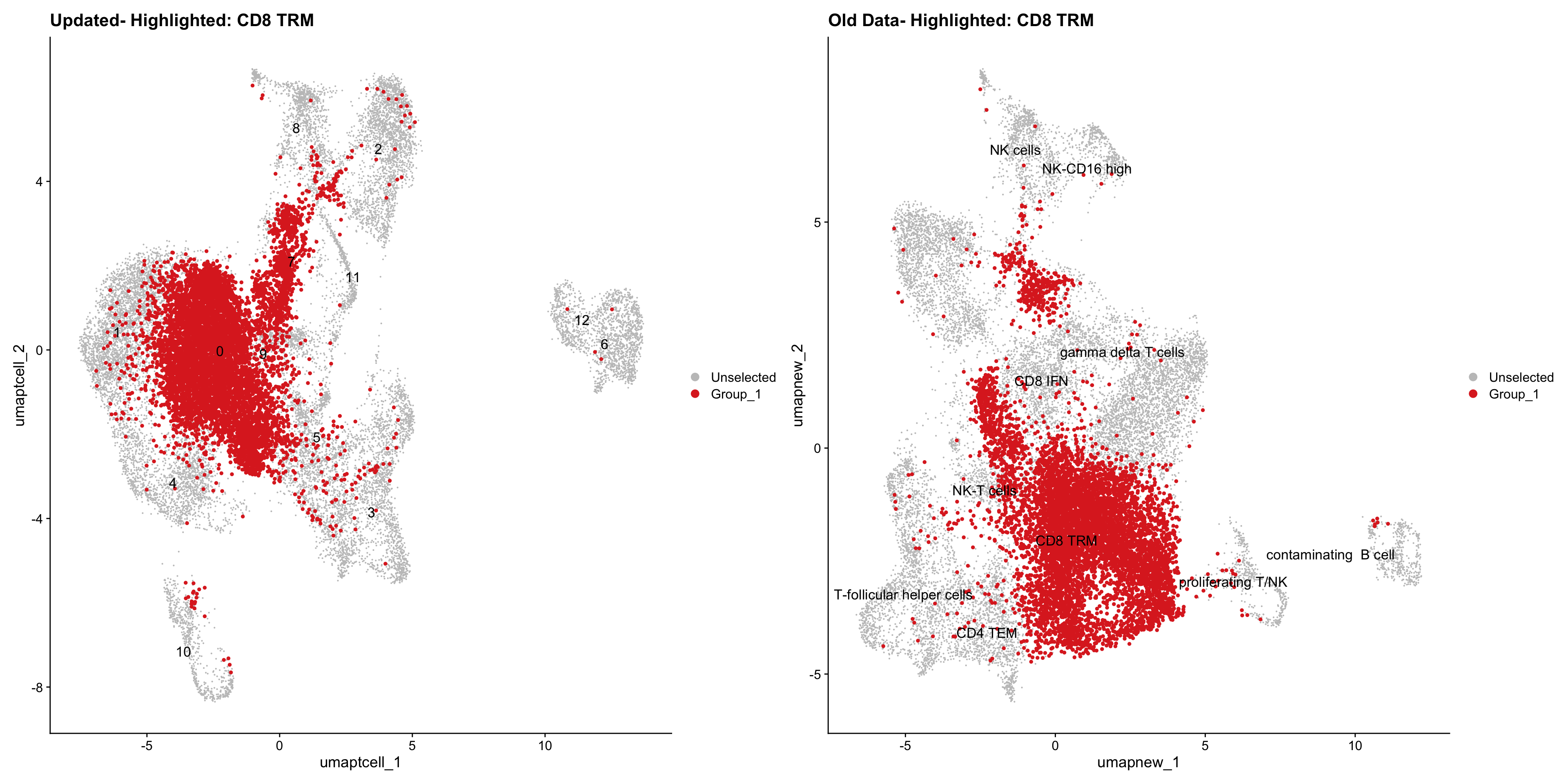
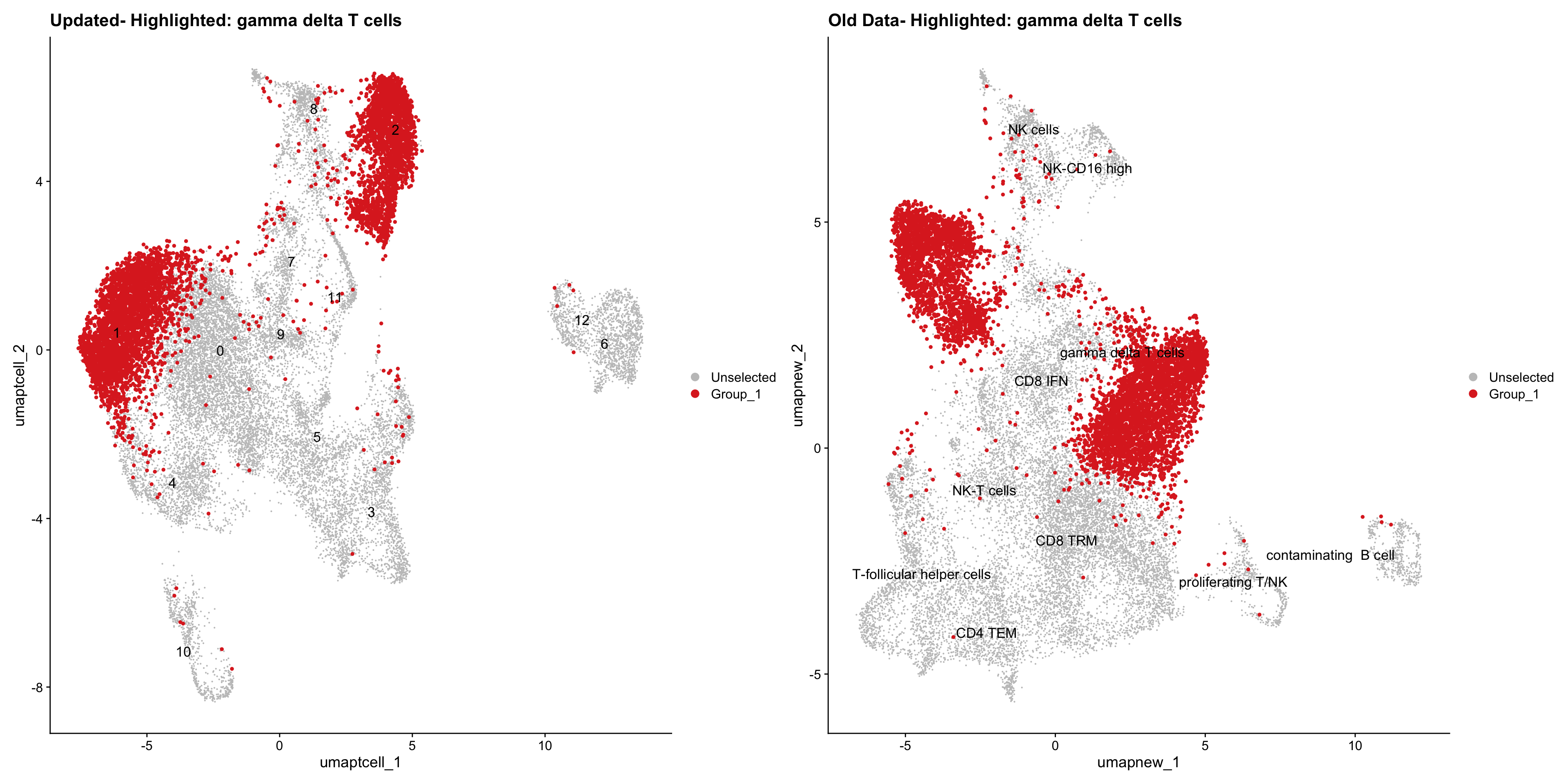
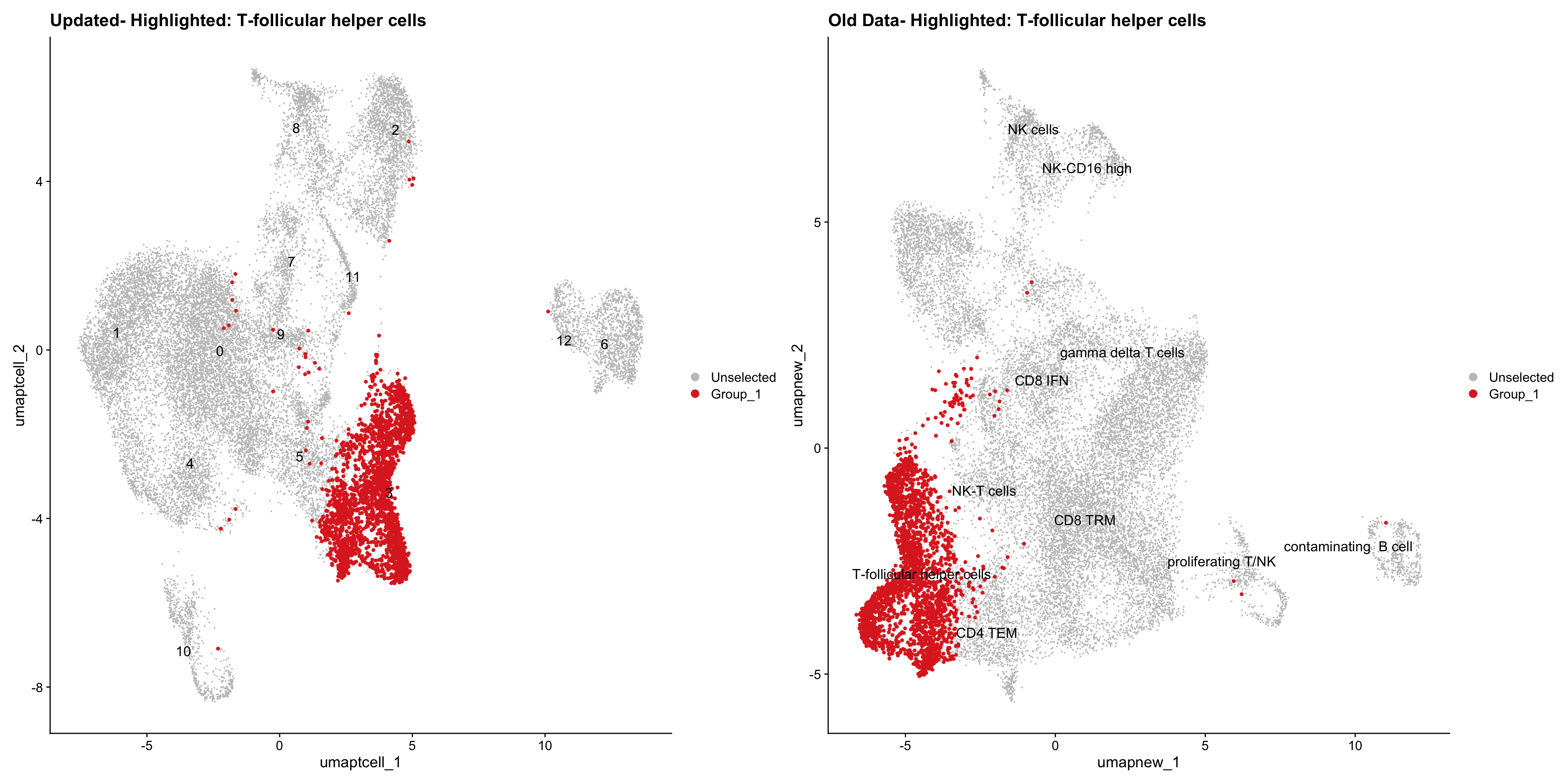
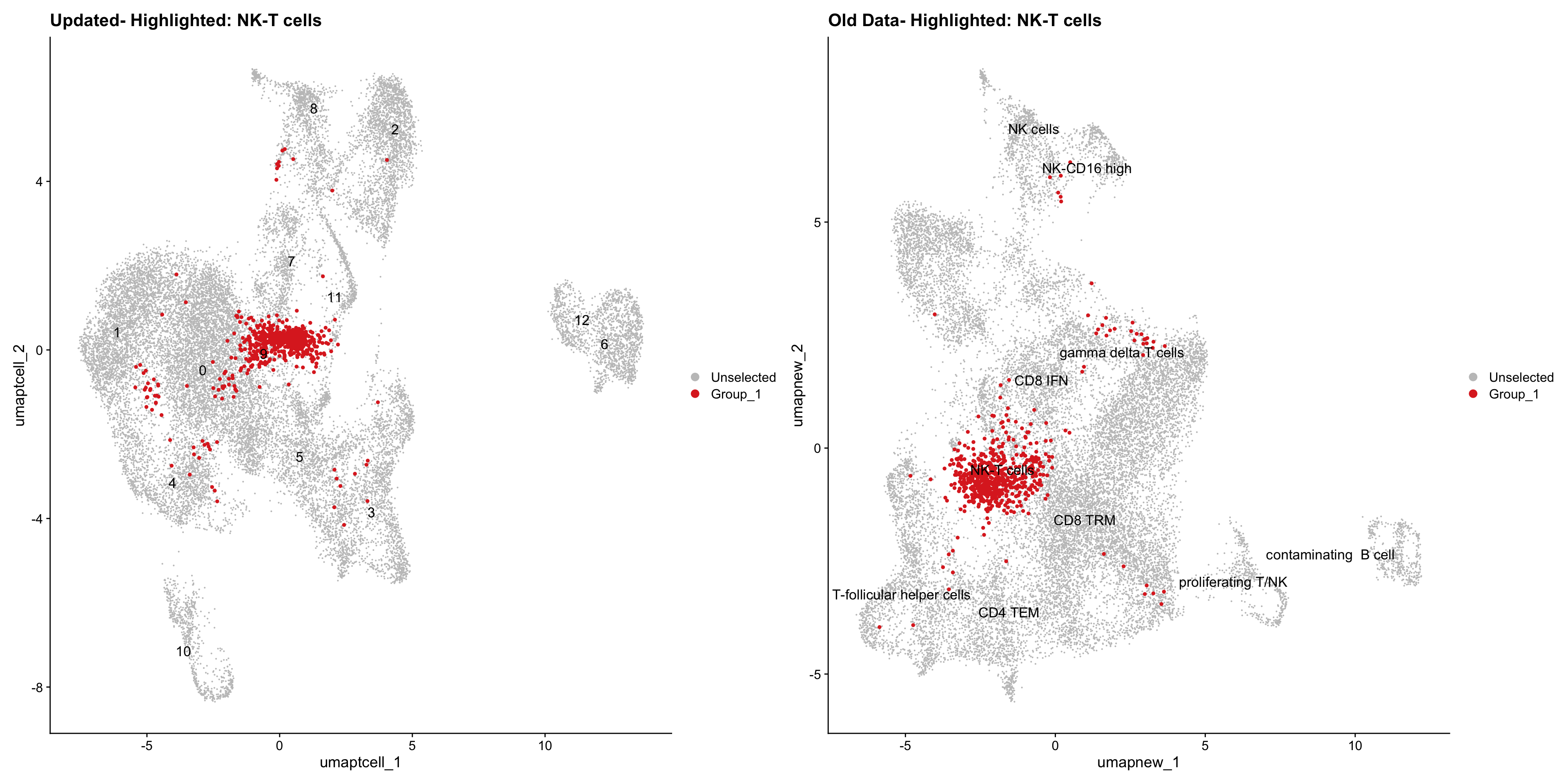
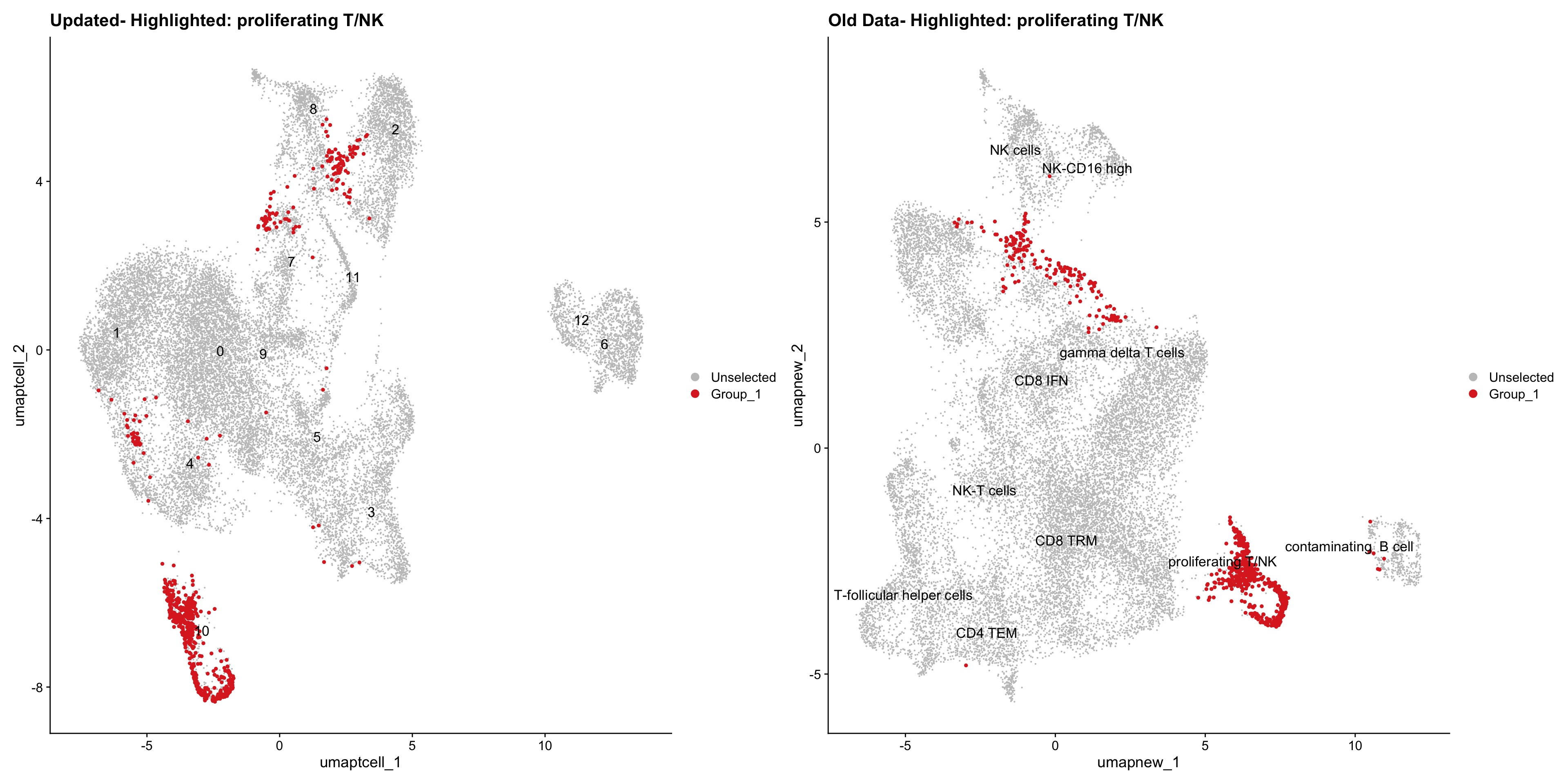
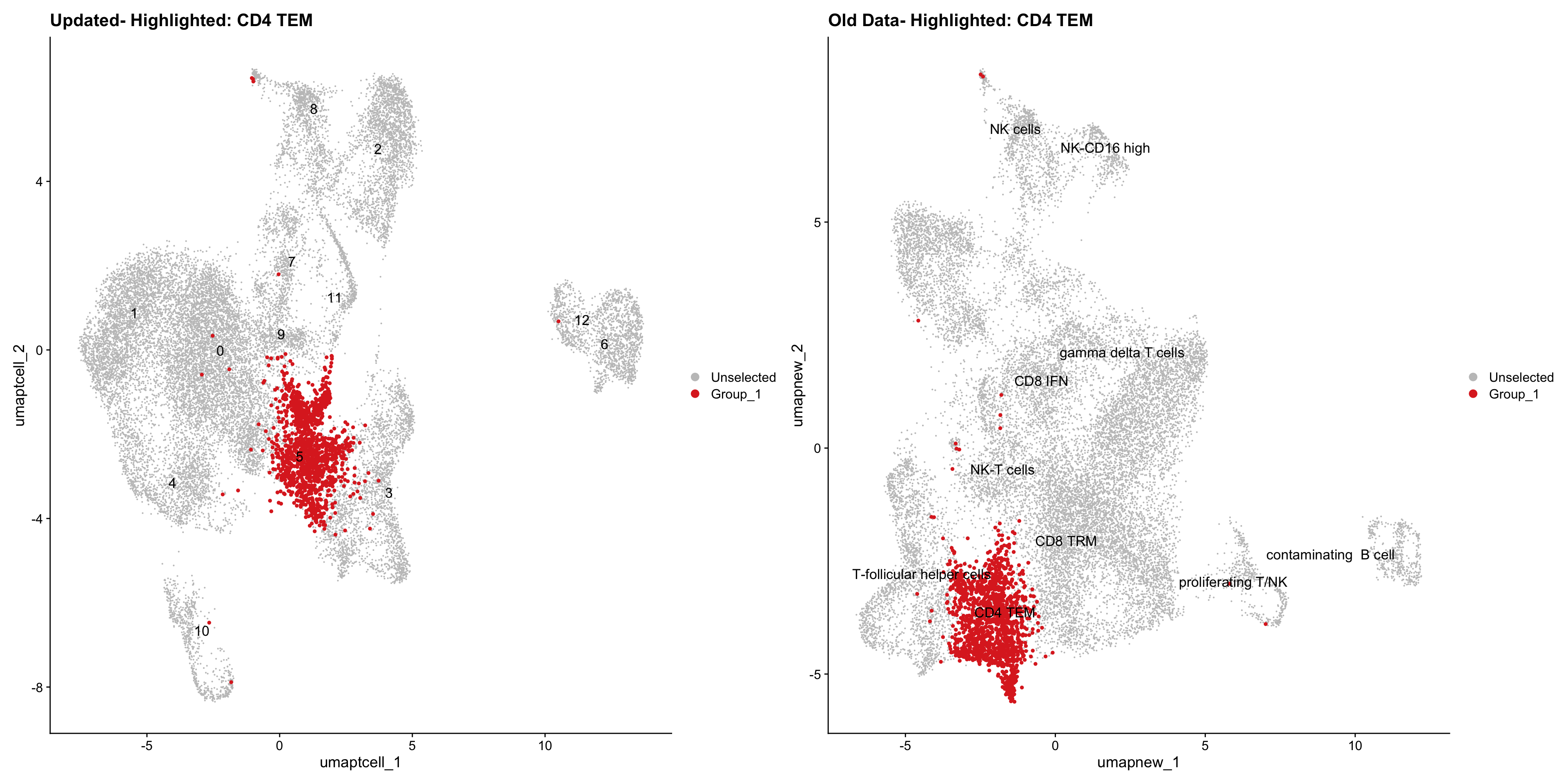
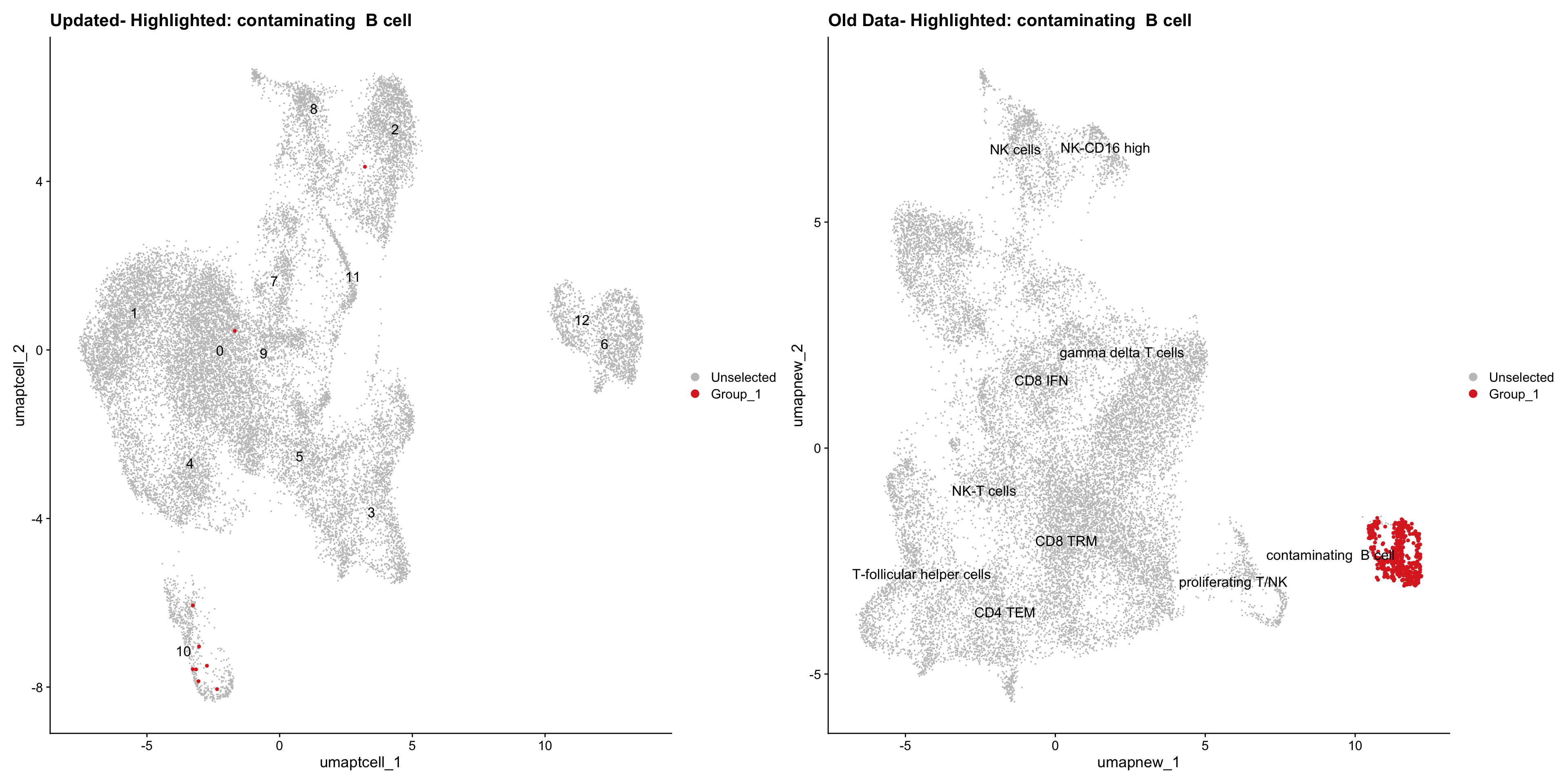
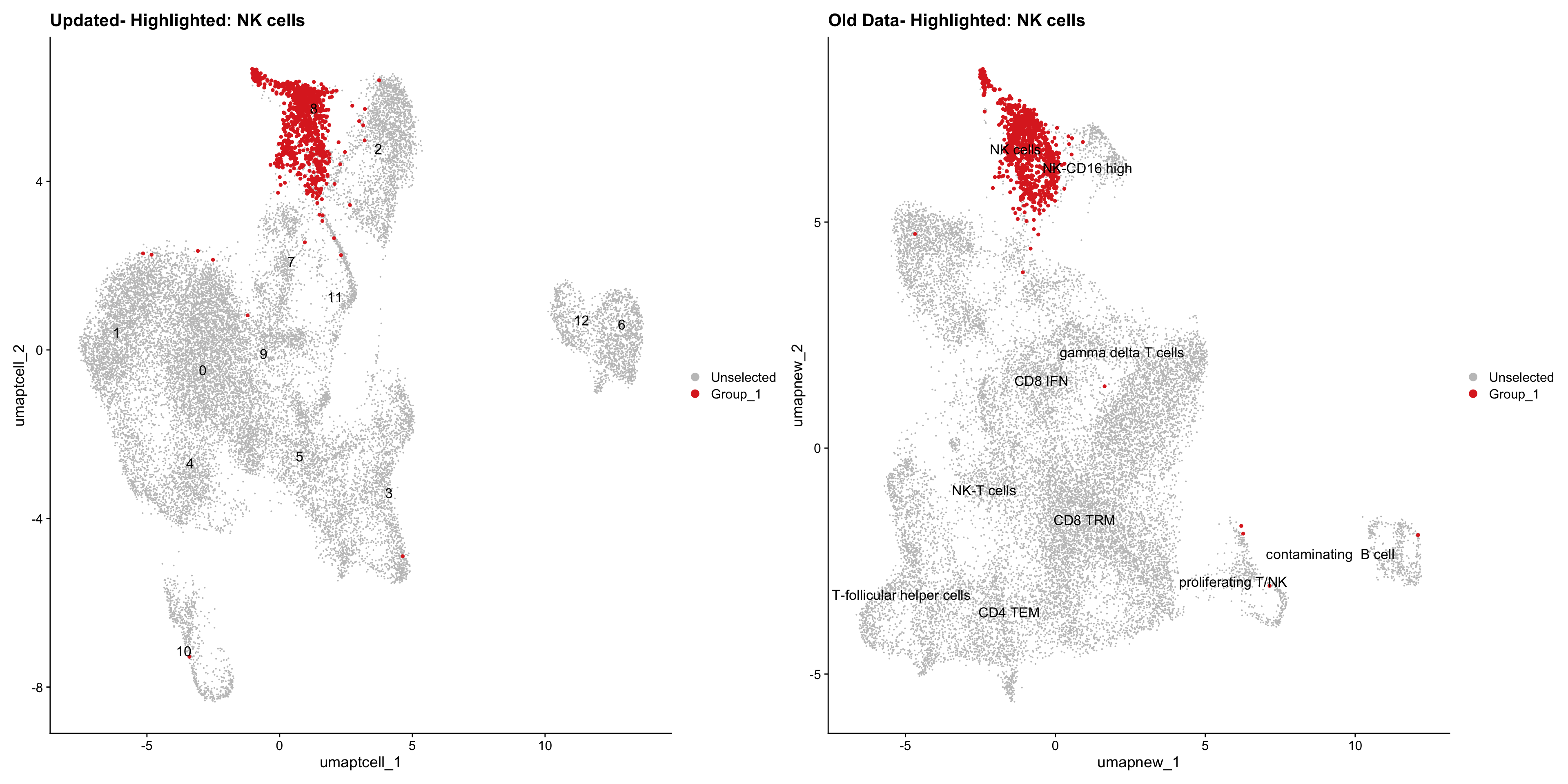
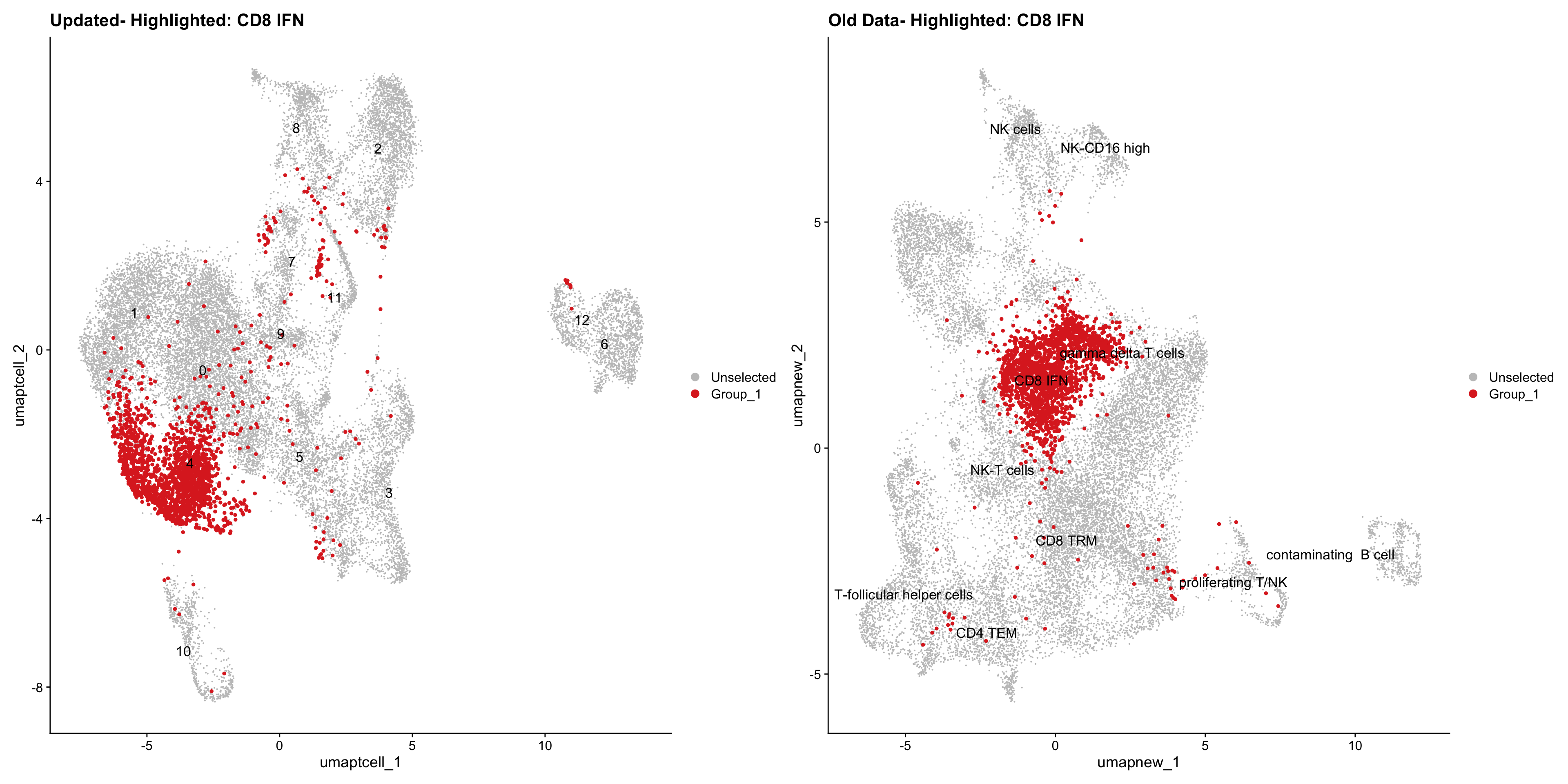
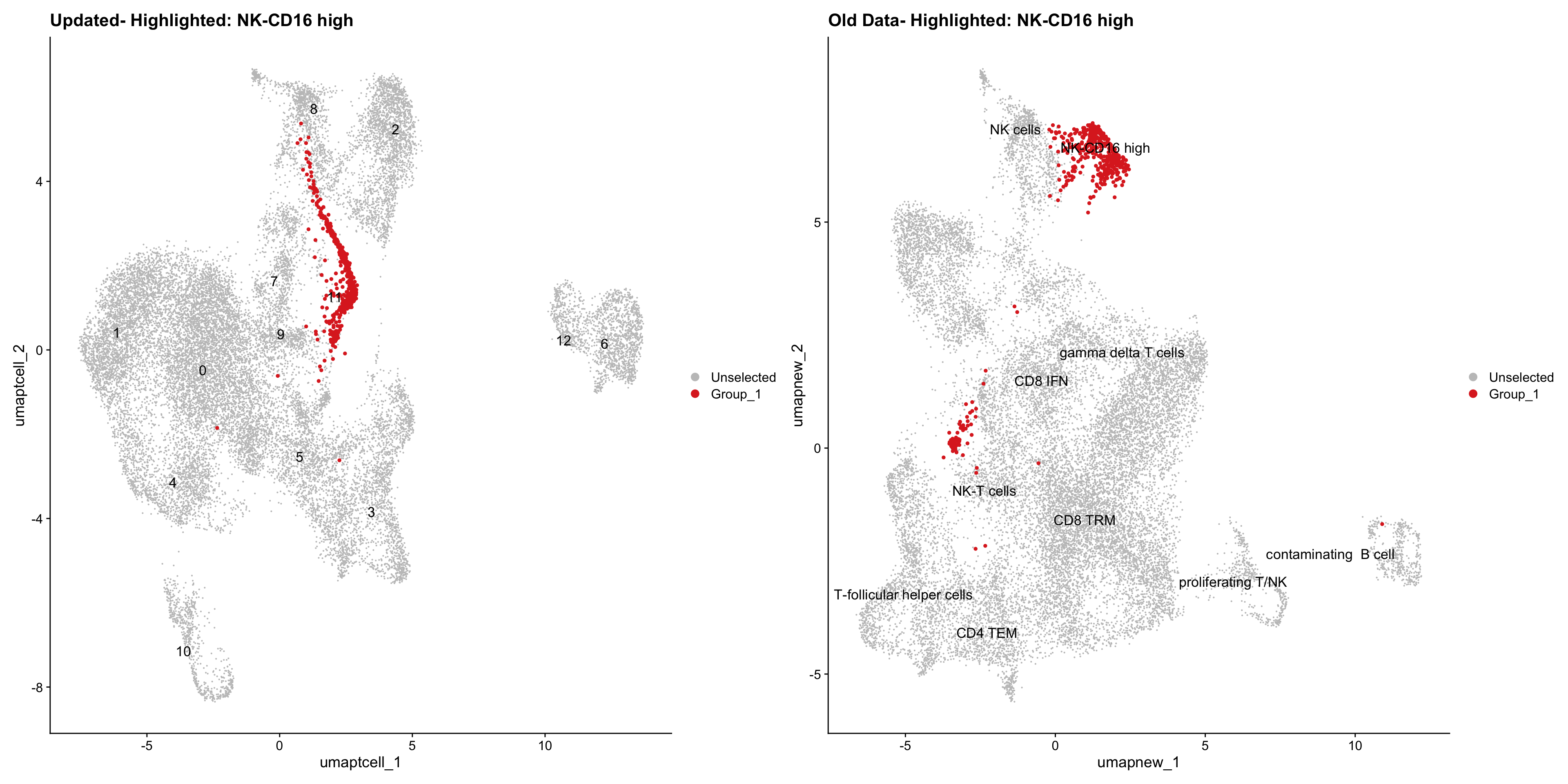
}Old data- cells from previous clusters higlighted
Loading old Subclustering seurat object of T cell population and comparing with the updated clustering.
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Tcell_population.subclusters.SEU.rds"))
old_obj <- readRDS(out2)cell_types <- unique(old_obj$cell_labels_v2)
for (cell_type in cell_types) {
cl_cells <- WhichCells(old_obj, idents = cell_type)
p <- DimPlot(
paed_sub,
reduction = "umap.tcell",
label = TRUE,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Updated- Highlighted:", cell_type))
p1 <- DimPlot(
old_obj,
reduction = "umap.new",
label = T,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Old Data- Highlighted:", cell_type))
print(p | p1)
}
palette1 <- paletteer::paletteer_d("ggthemes::Classic_20")
palette2 <- paletteer::paletteer_d("Polychrome::light")
combined_palette <- unique(c(palette1, palette2))
labels <- "RNA_snn_res.0.4"
p <- vector("list",length(labels))
for(label in labels){
paed_sub@meta.data %>%
ggplot(aes(x = !!sym(label),
fill = !!sym(label))) +
geom_bar() +
geom_text(aes(label = ..count..), stat = "count",
vjust = -0.5, colour = "black", size = 2) +
scale_y_log10() +
theme(axis.text.x = element_blank(),
axis.title.x = element_blank(),
axis.ticks.x = element_blank()) +
NoLegend() +
labs(y = "No. Cells (log scale)") -> p1
paed_sub@meta.data %>%
dplyr::select(!!sym(label), donor_id) %>%
group_by(!!sym(label), donor_id) %>%
summarise(num = n()) %>%
mutate(prop = num / sum(num)) %>%
ggplot(aes(x = !!sym(label), y = prop * 100,
fill = donor_id)) +
geom_bar(stat = "identity") +
theme(axis.text.x = element_text(angle = 90,
vjust = 0.5,
hjust = 1,
size = 8)) +
labs(y = "% Cells", fill = "Donor") +
scale_fill_manual(values = combined_palette) -> p2
(p1 / p2) & theme(legend.text = element_text(size = 8),
legend.key.size = unit(3, "mm")) -> p[[label]]
}`summarise()` has grouped output by 'RNA_snn_res.0.4'. You can override using
the `.groups` argument.p[[1]]
NULL
$RNA_snn_res.0.4Warning: The dot-dot notation (`..count..`) was deprecated in ggplot2 3.4.0.
ℹ Please use `after_stat(count)` instead.
This warning is displayed once every 8 hours.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.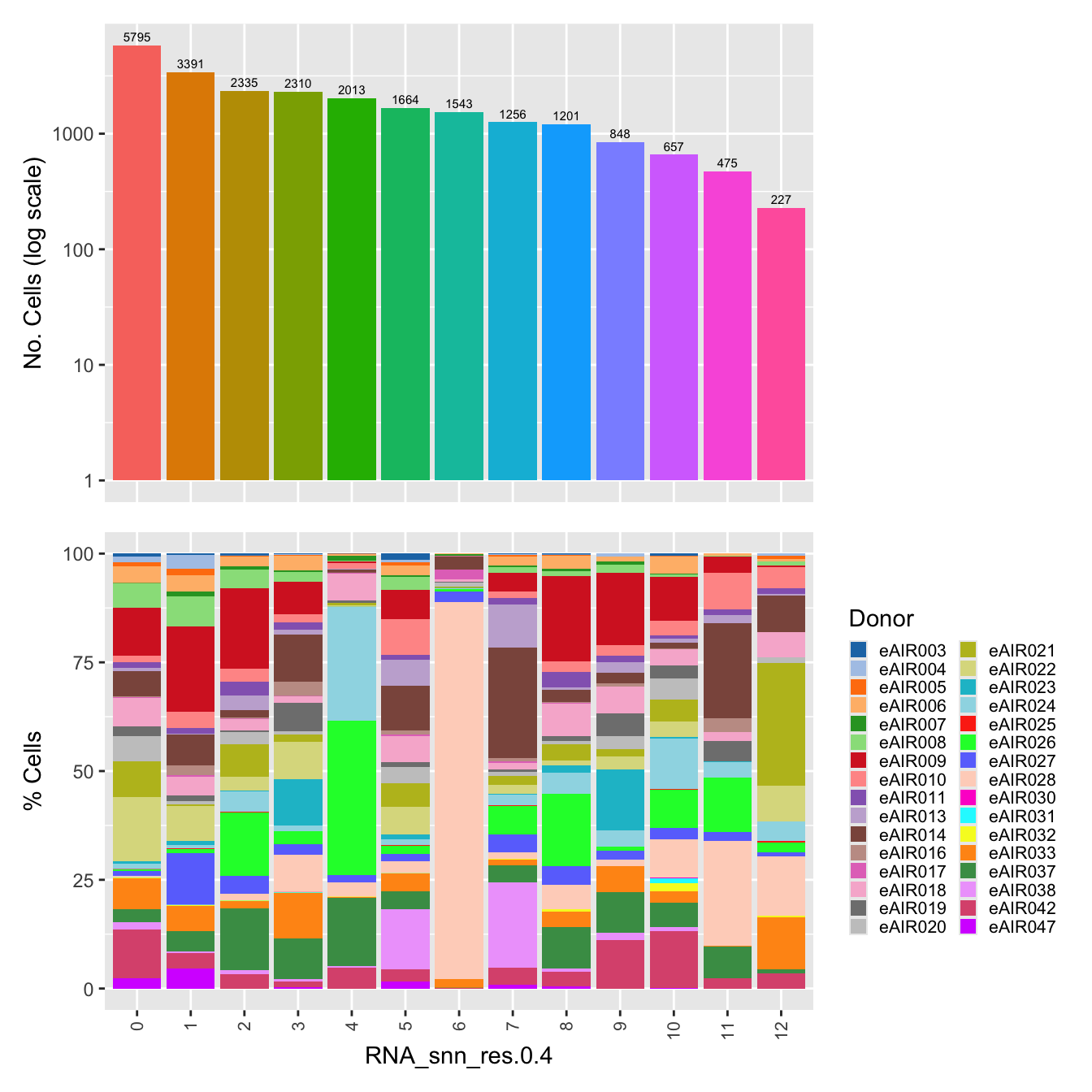
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
Save subclustered SEU object (Tcells)
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs_v2", tissue,
paste0("G000231_Neeland_",tissue,".Tcell_population.subclusters.SEU.rds"))
#dir.create(out2)
#if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
#}Updated cell-type labels (T cell clusters)
cell_labels <- readxl::read_excel(here("data/cell_labels_Mel_v4_Dec2024/earlyAIR_NB_all.xlsx"), sheet = "T-reclustering")
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
paed_sub <- RenameIdents(paed_sub, new_cluster_names)
paed_sub@meta.data$cell_labels_v2 <- Idents(paed_sub)
p3 <- DimPlot(paed_sub, reduction = "umap.tcell", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated T cell population"))
p3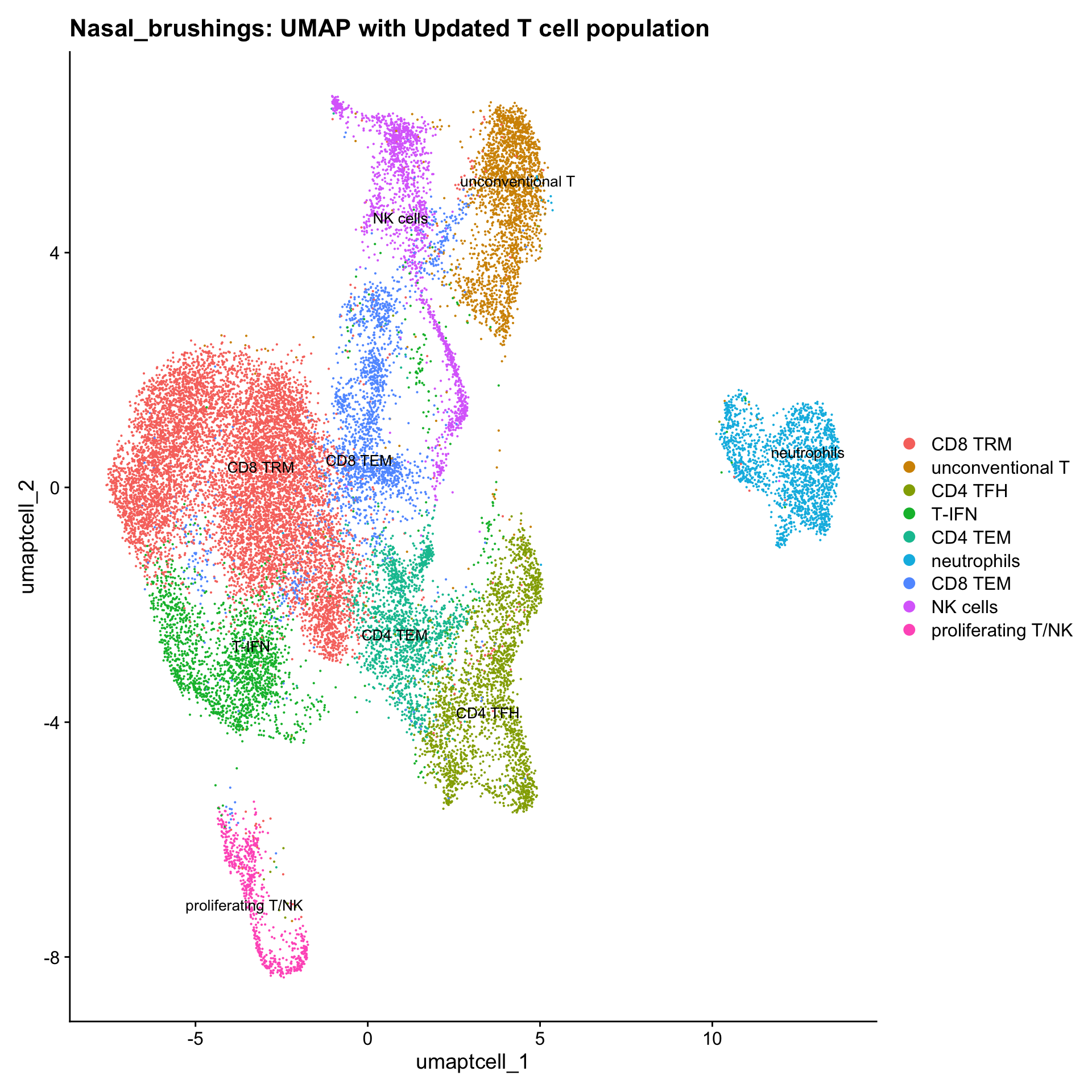
Save subclustered SEU object
out2 <- here("output",
"RDS", "AllBatches_Annotated_Subclustering_SEUs_v2", tissue,
paste0("G000231_Neeland_",tissue,".Tcell_population.subclusters.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Reclustering B cell population
The marker genes for this reclustering can be found here-
idx <- which(Idents(merged_obj) %in% "B cells for reclustering")
paed_bcells <- merged_obj[,idx]
mito_genes <- grep("^MT-", rownames(paed_bcells), value = TRUE)
paed_bcells <- subset(paed_bcells, features = setdiff(rownames(paed_bcells), mito_genes))
paed_bcellsAn object of class Seurat
17962 features across 12924 samples within 1 assay
Active assay: RNA (17962 features, 2000 variable features)
3 layers present: data, counts, scale.data
4 dimensional reductions calculated: pca, umap.unintegrated, harmony, umap.harmonypaed_bcells <- paed_bcells %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_bcells <- RunUMAP(paed_bcells, dims = 1:30, reduction = "pca", reduction.name = "umap.bcell")
meta_data_columns <- colnames(paed_bcells@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_bcells@meta.data <- paed_bcells@meta.data[, !(colnames(paed_bcells@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_bcells <- FindNeighbors(paed_bcells, reduction = "pca", dims = 1:30)
paed_bcells <- FindClusters(paed_bcells, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9393
Number of communities: 6
Elapsed time: 9 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9120
Number of communities: 9
Elapsed time: 7 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8938
Number of communities: 10
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8795
Number of communities: 10
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8653
Number of communities: 11
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8520
Number of communities: 12
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8400
Number of communities: 12
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8284
Number of communities: 13
Elapsed time: 6 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8174
Number of communities: 13
Elapsed time: 5 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 12924
Number of edges: 425959
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8074
Number of communities: 14
Elapsed time: 5 secondsDimHeatmap(paed_bcells, dims = 1:10, cells = 500, balanced = TRUE)
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
clustree(paed_bcells, prefix = "RNA_snn_res.")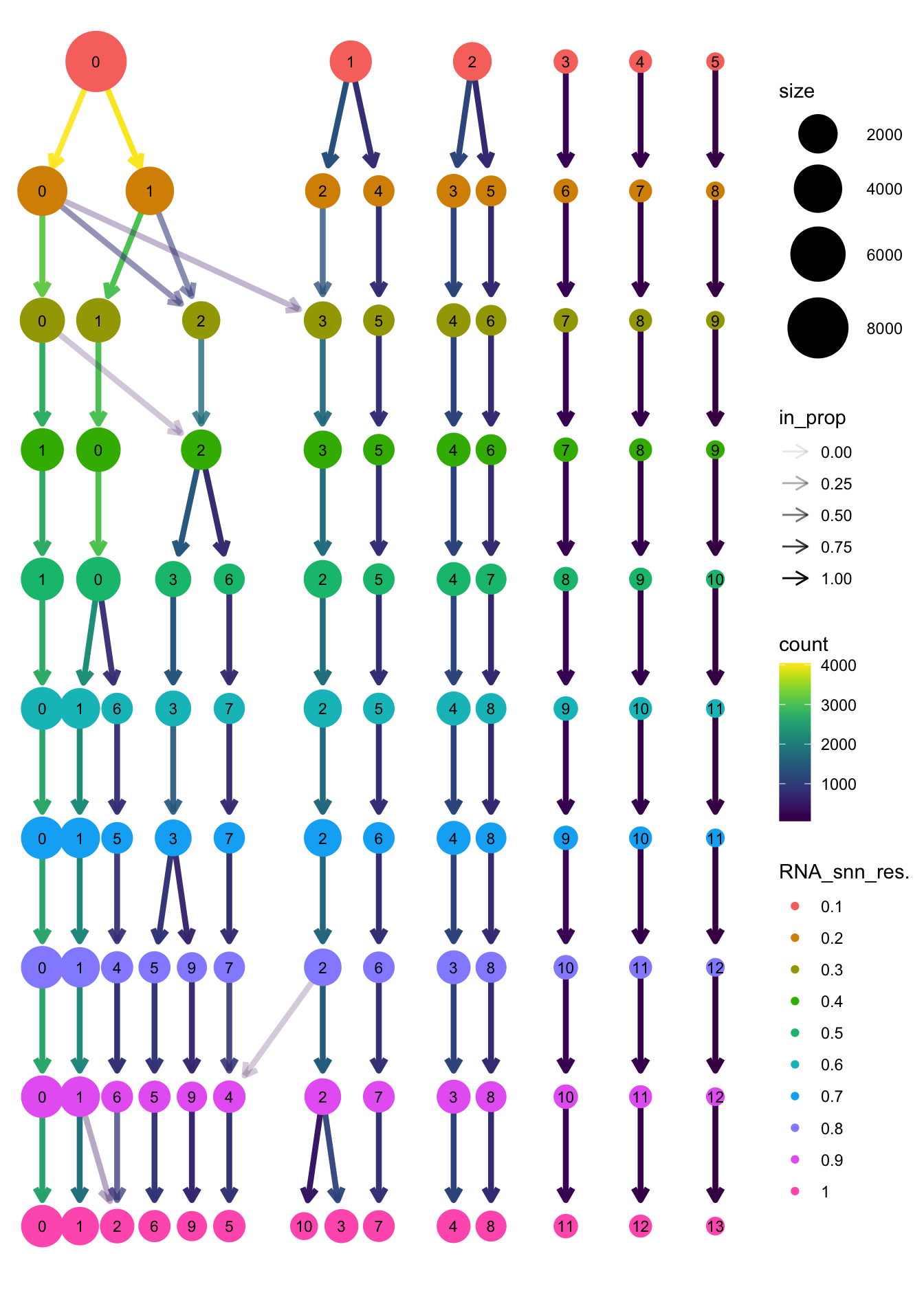
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
opt_res <- "RNA_snn_res.0.2"
n <- nlevels(paed_bcells$RNA_snn_res.0.2)
paed_bcells$RNA_snn_res.0.2 <- factor(paed_bcells$RNA_snn_res.0.2, levels = seq(0,n-1))
paed_bcells$seurat_clusters <- NULL
Idents(paed_bcells) <- paed_bcells$RNA_snn_res.0.2DimPlot(paed_bcells,reduction = "umap.bcell" ,group.by = "RNA_snn_res.0.2", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )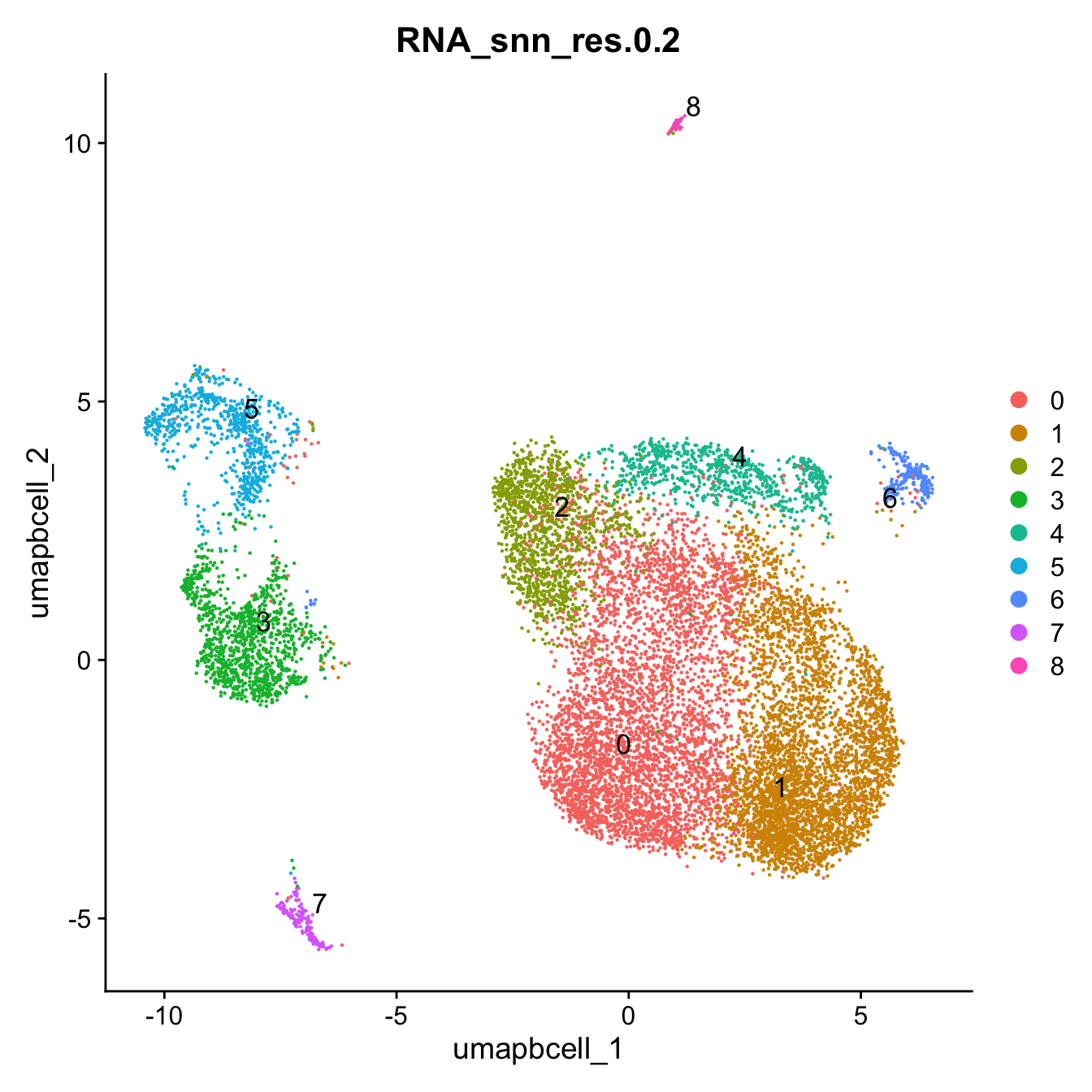
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
paed_bcells <- JoinLayers(paed_bcells)
paed_bcells.markers <- FindAllMarkers(paed_bcells, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8paed_bcells.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_bcells.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster[1] "B2M" "IGHD" "ITGAX" "MEF2B" "DUSP1" "MYBL2" "G0S2" "MZB1" "CD2" FeaturePlot(paed_bcells,features=best.wilcox.gene.per.cluster, reduction="umap.bcell" ,raster = FALSE, label = T, ncol = 3)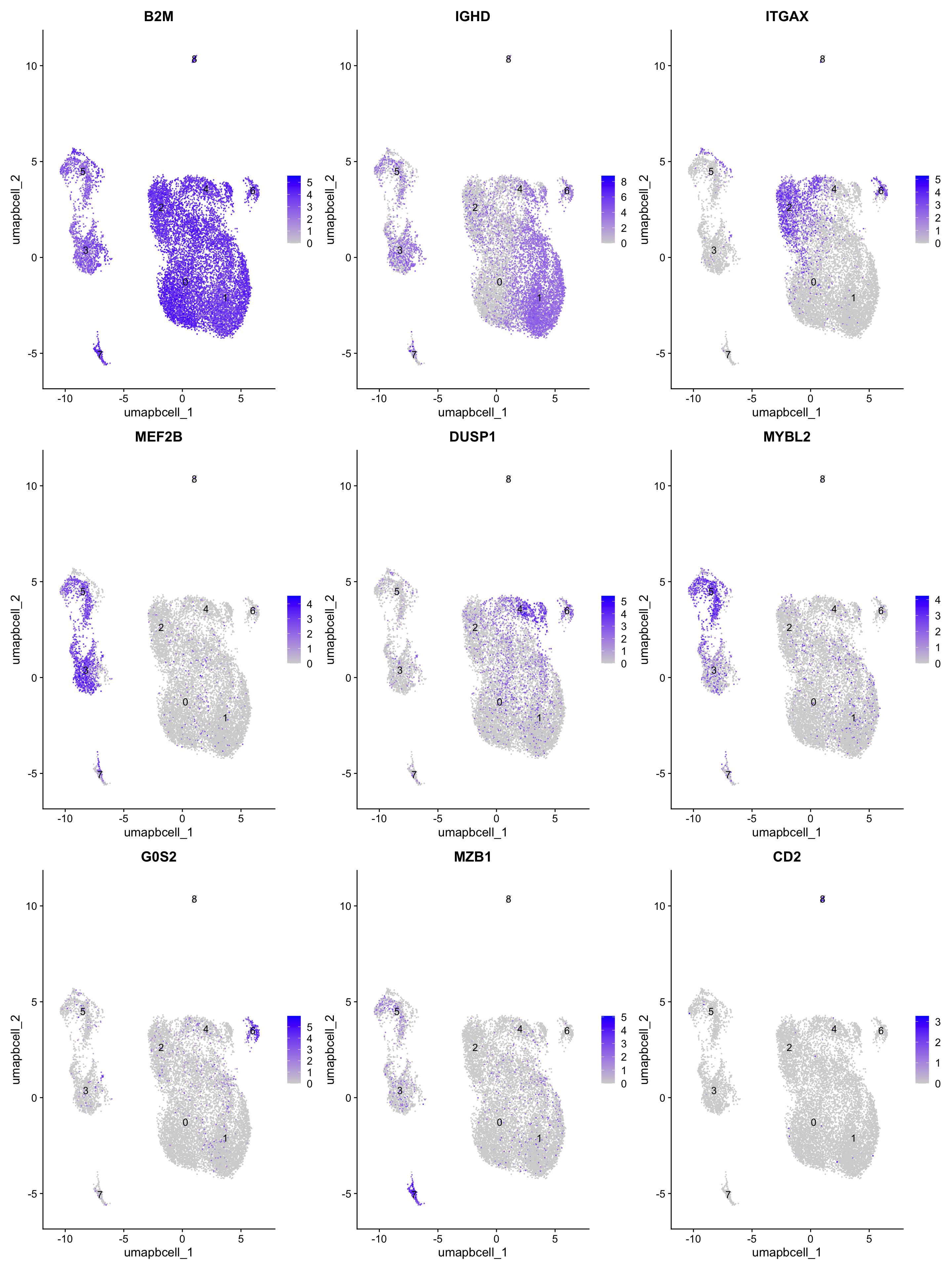
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_bcells.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_bcells,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.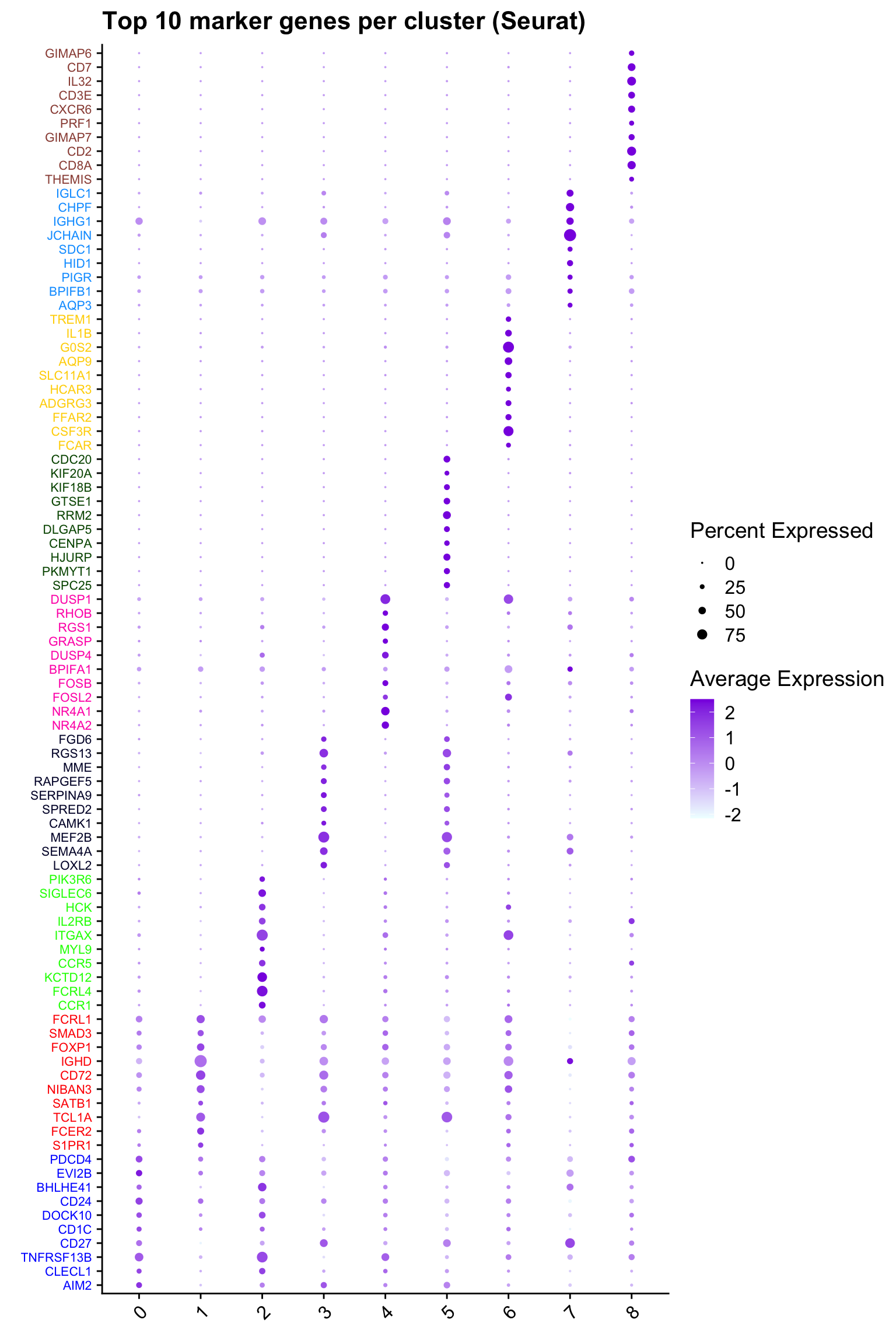
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
out_markers <- here("output",
"CSV_v2",tissue,
paste(tissue,"_Marker_genes_Reclustered_Bcell_population.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_bcells.markers$cluster)) {
cluster_data <- paed_bcells.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
if (!file.exists(file_name)) {
write.csv(cluster_data, file = file_name)
}
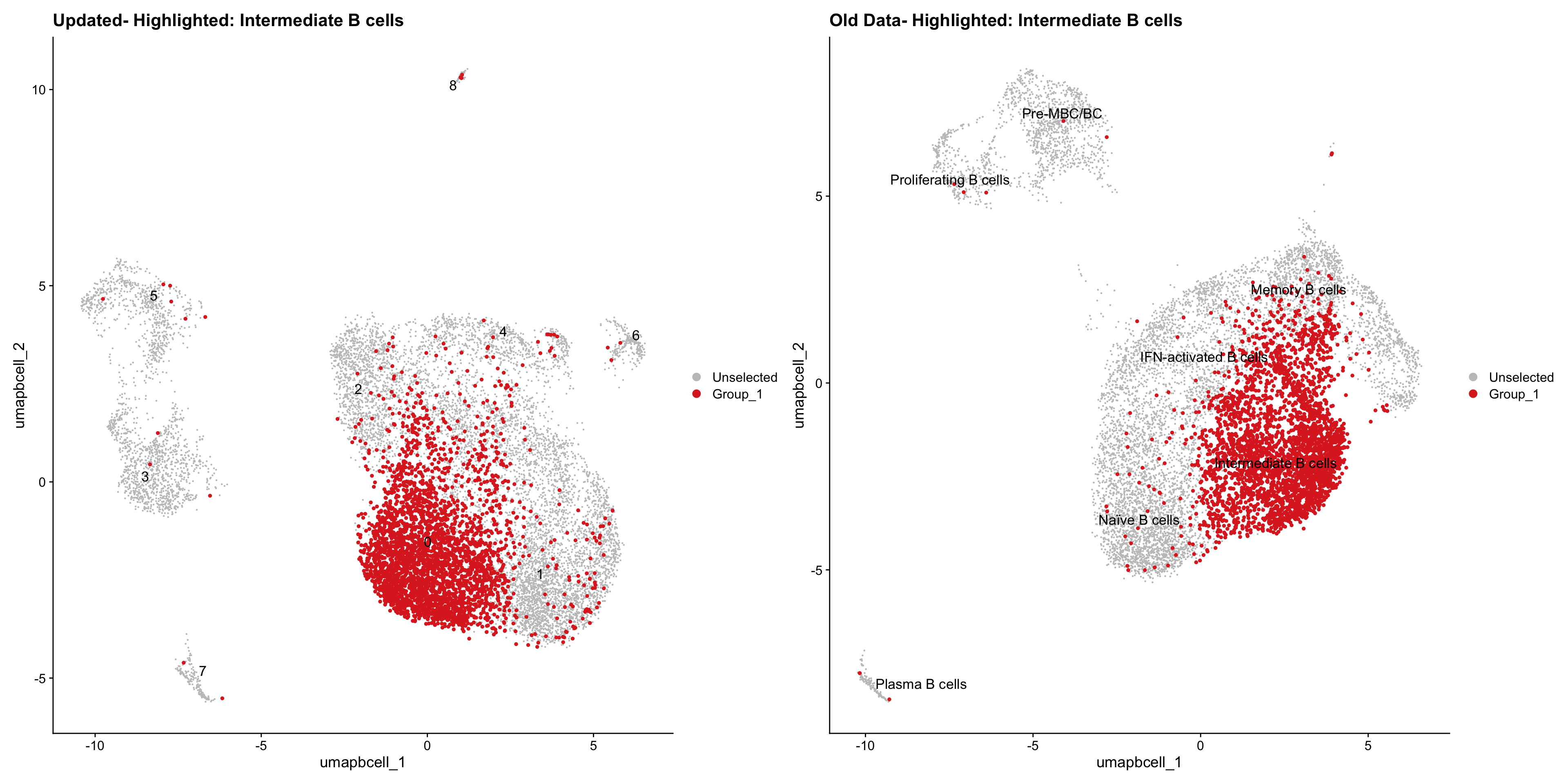
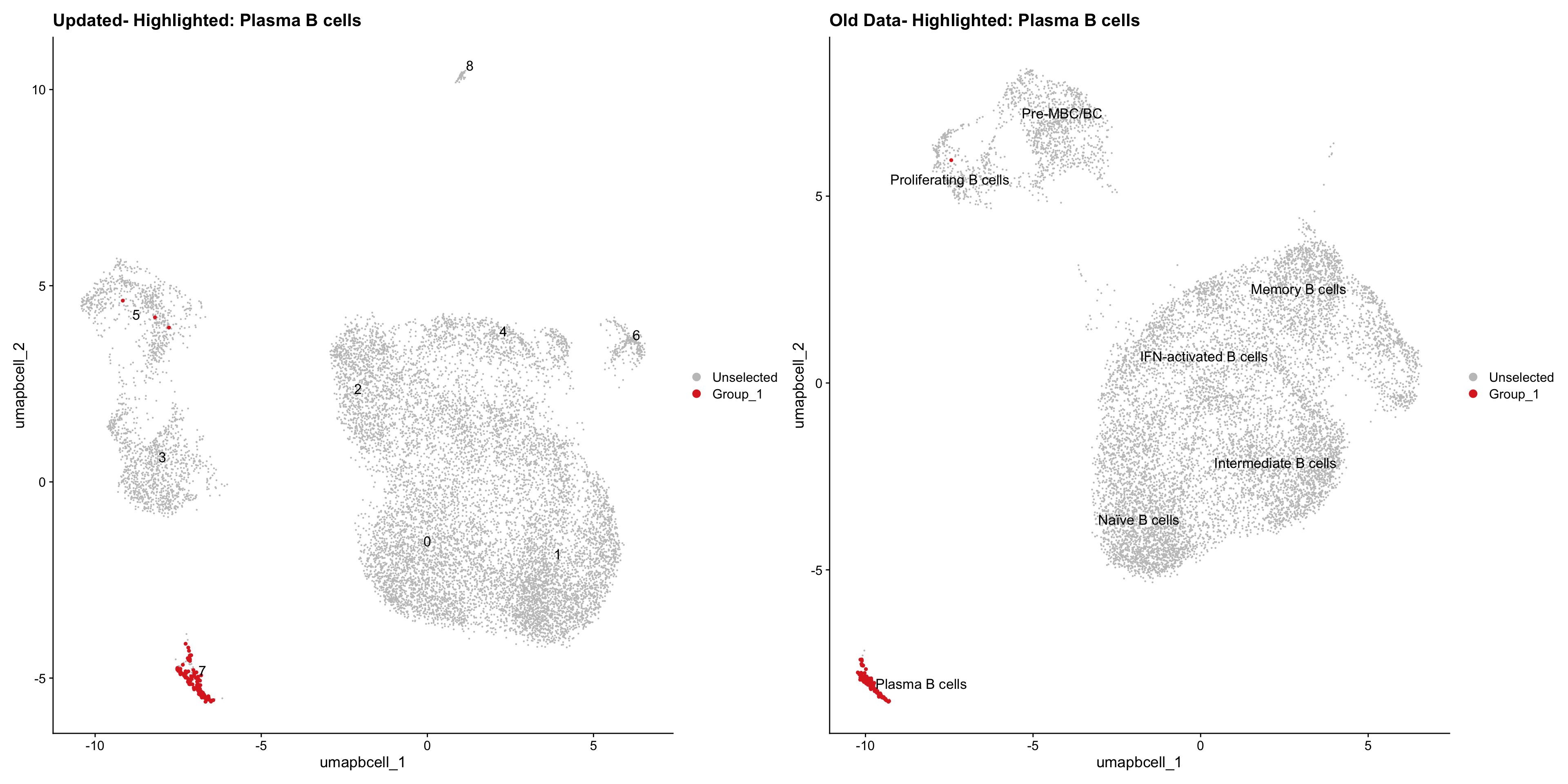
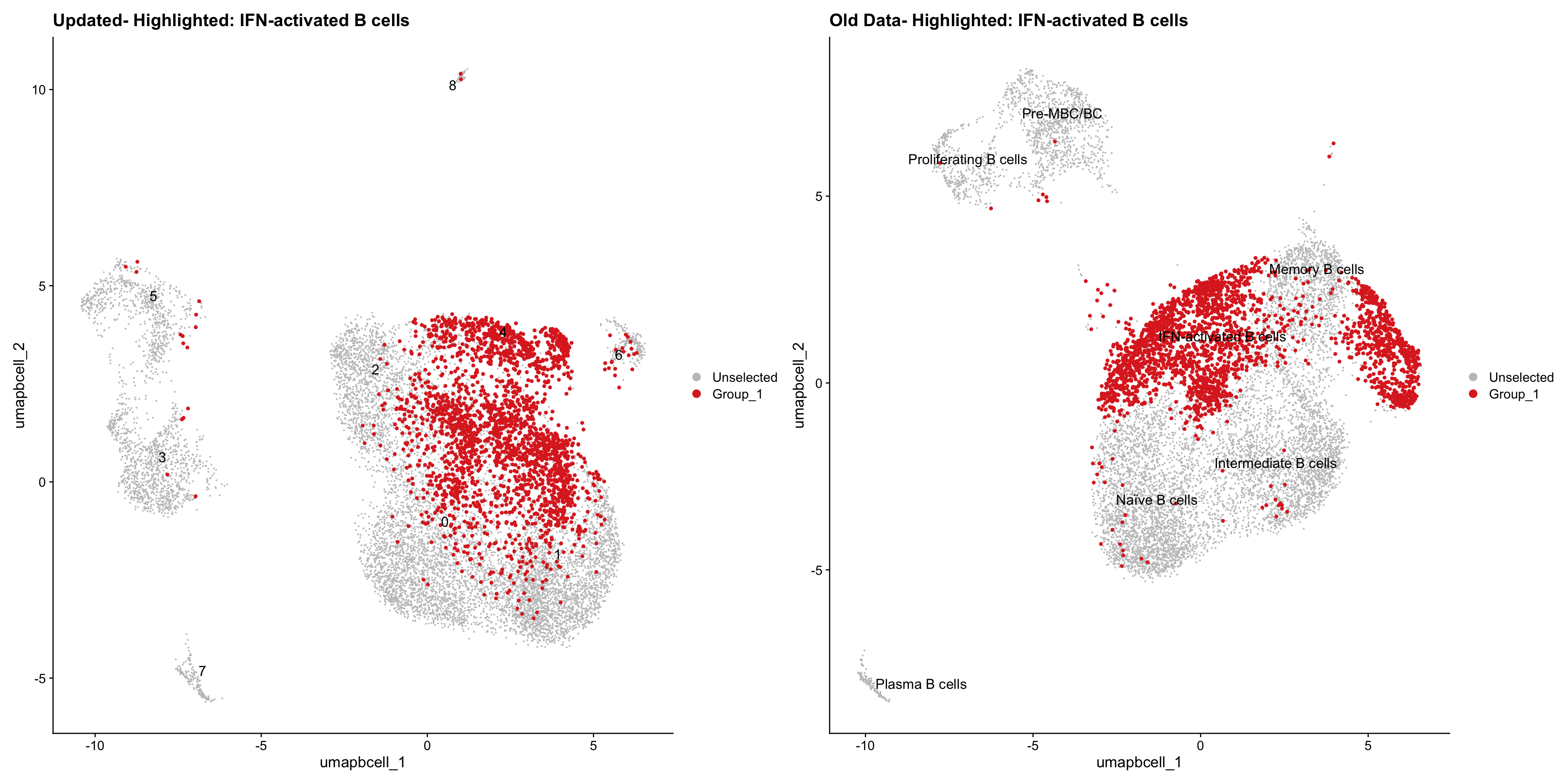
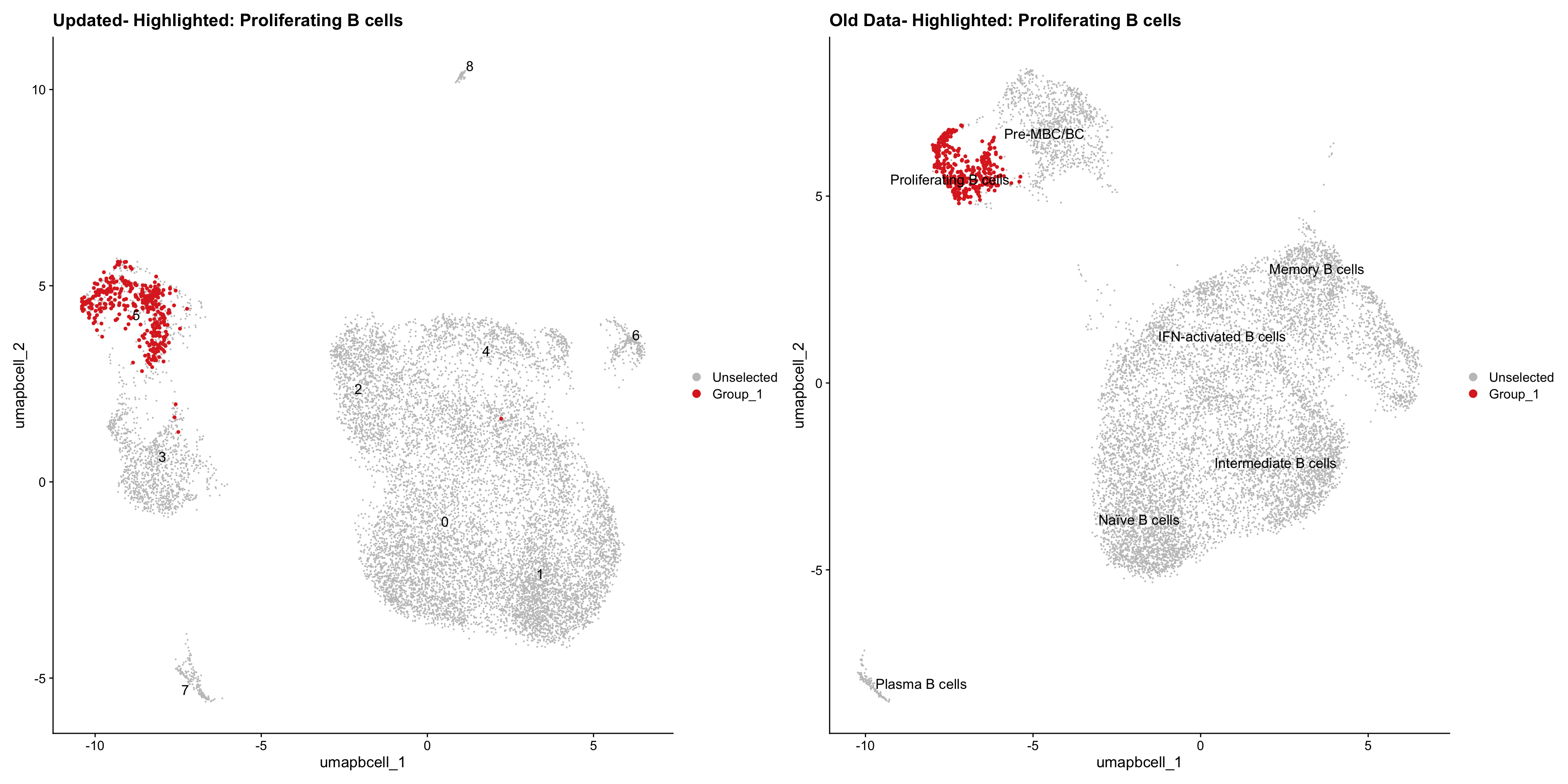
}Old data- cells from previous clusters higlighted
Loading old Subclustering seurat object of T cell population and comparing with the updated clustering.
out2 <- here("output",
"RDS","AllBatches_Subclustering_SEUs", "AllBatches_Subclustering_v2_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Bcell_population.subclusters.SEU.rds"))
old_obj <- readRDS(out2)cell_types <- unique(old_obj$cell_labels_v2)
for (cell_type in cell_types) {
cl_cells <- WhichCells(old_obj, idents = cell_type)
p <- DimPlot(
paed_bcells,
reduction = "umap.bcell",
label = TRUE,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Updated- Highlighted:", cell_type))
p1 <- DimPlot(
old_obj,
reduction = "umap.bcell",
label = T,
label.size = 4.5,
repel = TRUE,
raster = FALSE,
cells.highlight = cl_cells
) +
ggtitle(paste("Old Data- Highlighted:", cell_type))
print(p | p1)
}
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
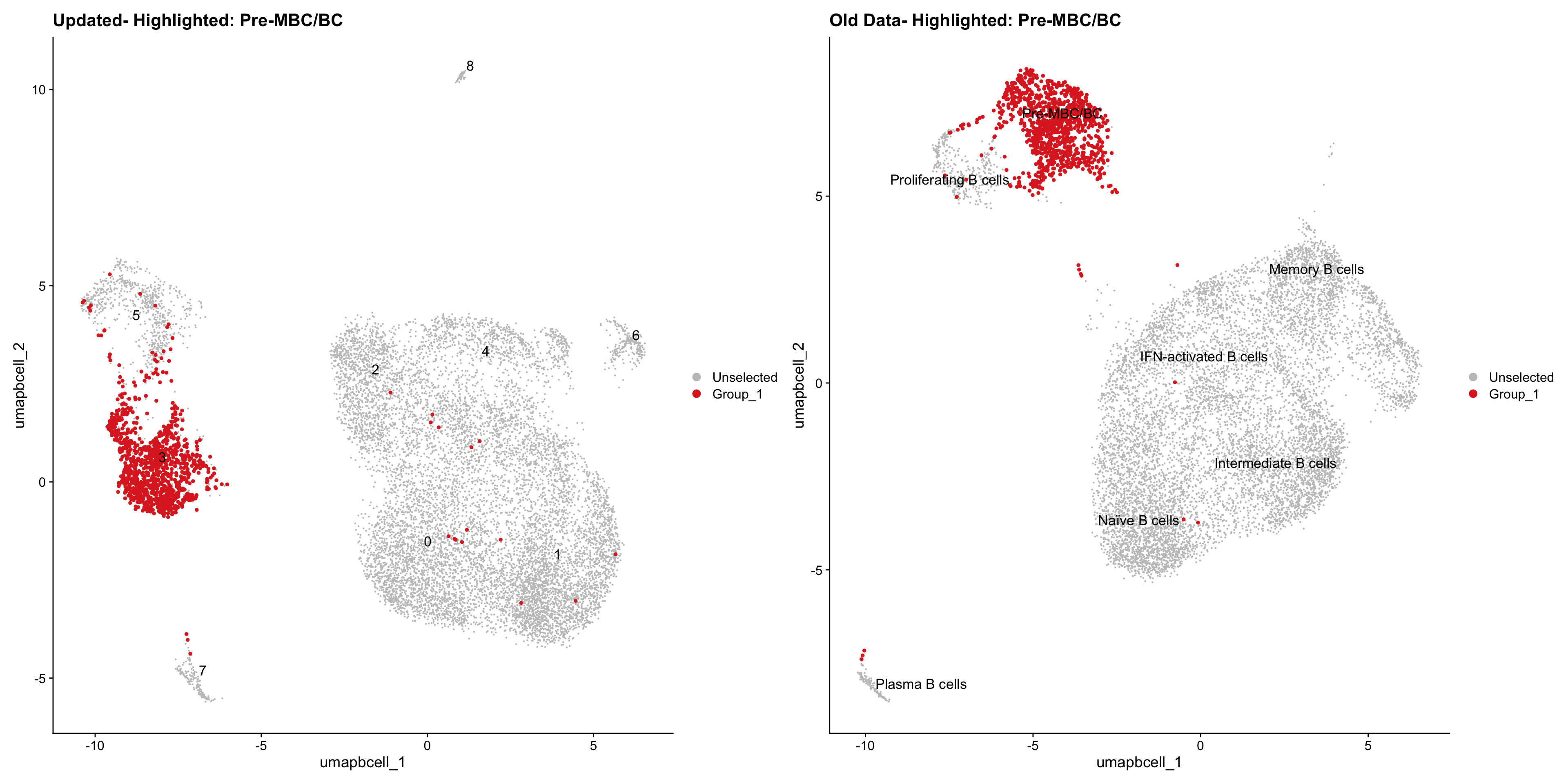
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
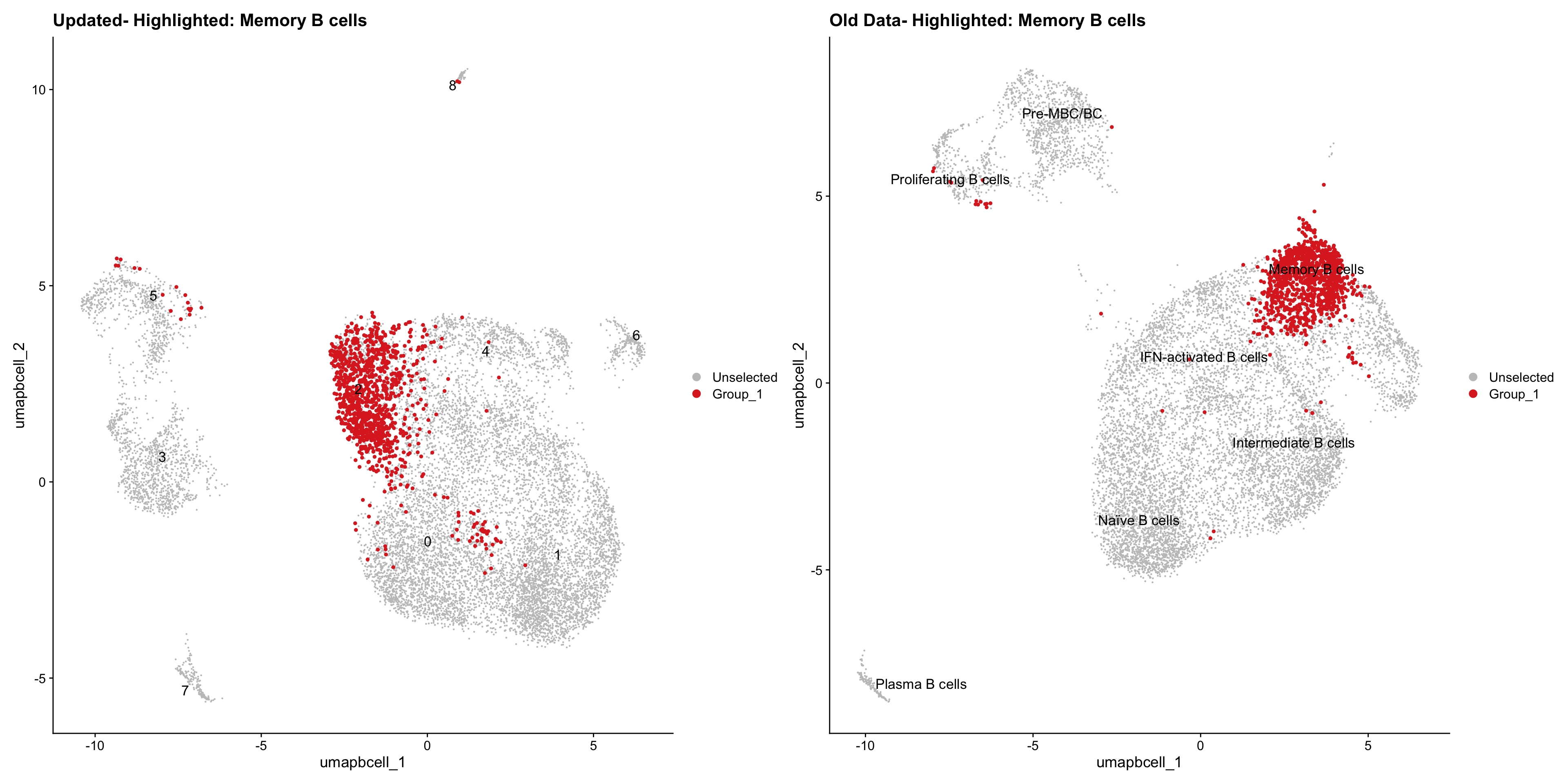
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
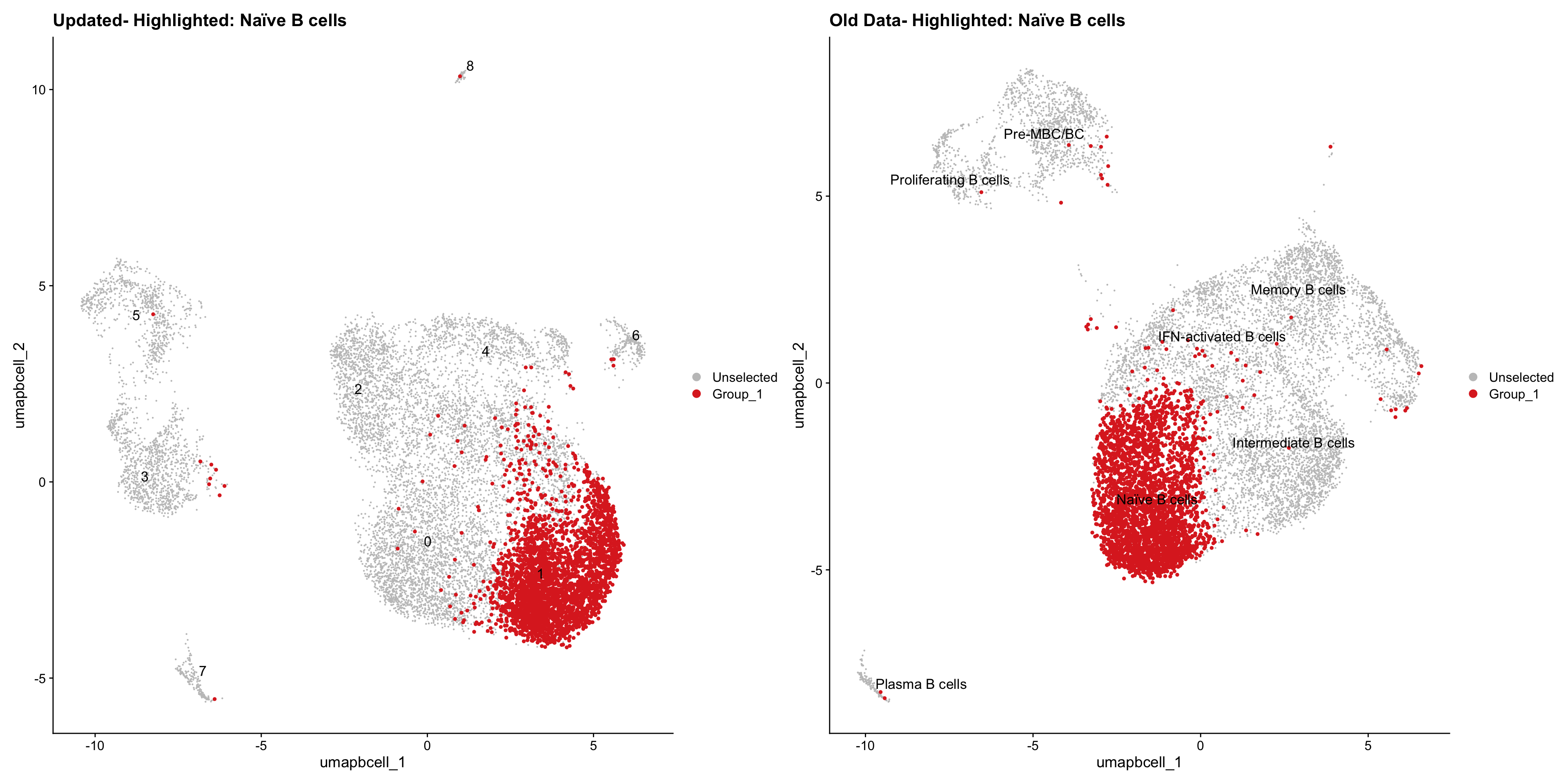
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
| Version | Author | Date |
|---|---|---|
| 3595ad0 | Gunjan Dixit | 2025-01-07 |
Updated cell-type labels (B cell clusters)
cell_labels <- readxl::read_excel(here("data/cell_labels_Mel_v4_Dec2024/earlyAIR_NB_all.xlsx"), sheet = "B-reclustering")
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
paed_bcells <- RenameIdents(paed_bcells, new_cluster_names)
paed_bcells@meta.data$cell_labels_v2 <- Idents(paed_bcells)
p3 <- DimPlot(paed_bcells, reduction = "umap.bcell", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated B cell population"))
p3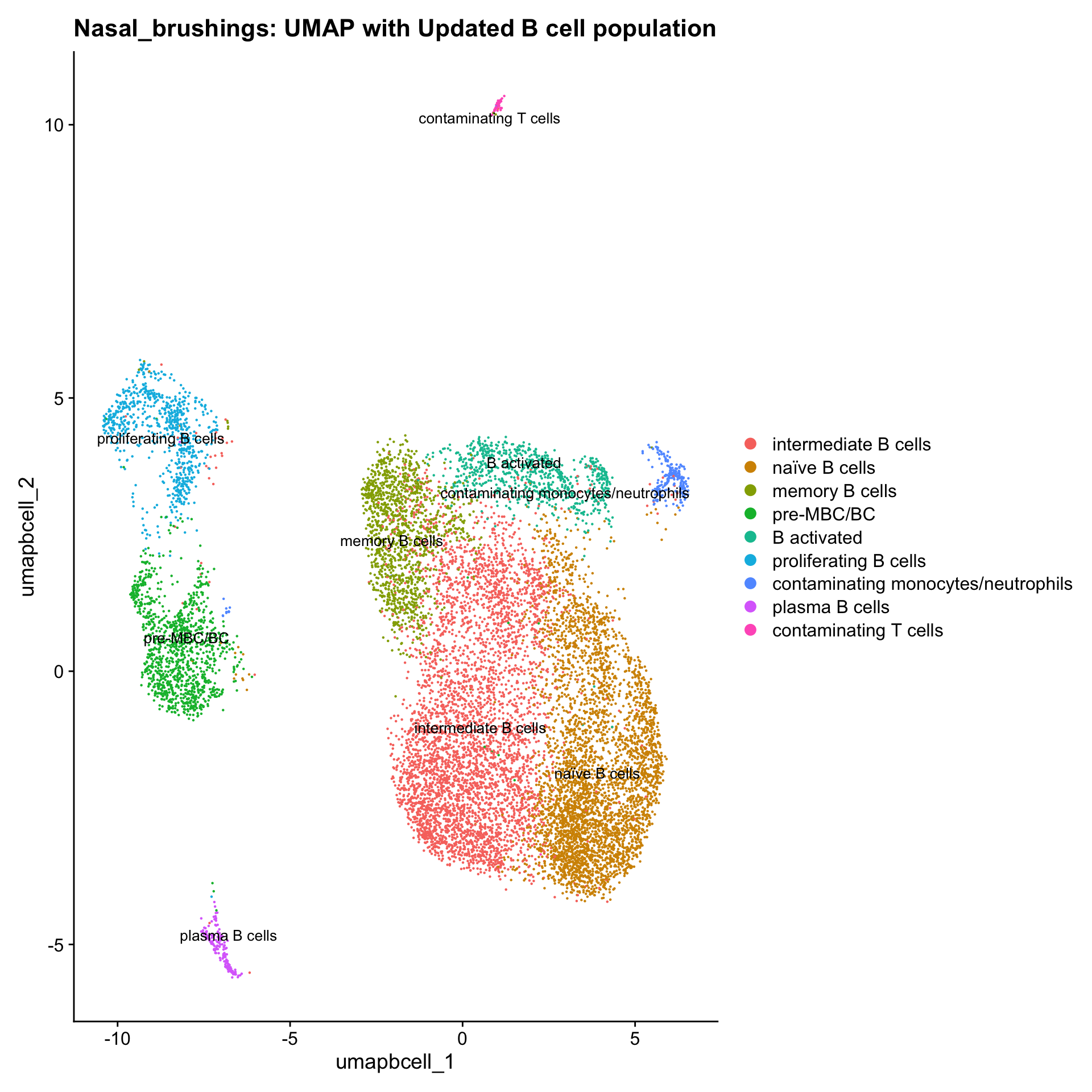
table(paed_bcells$cell_labels_v2)
intermediate B cells naïve B cells
4446 4019
memory B cells pre-MBC/BC
1334 1134
B activated proliferating B cells
802 748
contaminating monocytes/neutrophils plasma B cells
226 156
contaminating T cells
59 Since there are only 59 contaminated T cells here, I will chuck them out.
Excluding contaminating labels
idx <- which(grepl("^contaminating", Idents(paed_bcells)))
paed_bcells <- paed_bcells[, -idx]Save subclustered SEU object Bcells
out2 <- here("output",
"RDS", "AllBatches_Annotated_Subclustering_SEUs_v2", tissue,
paste0("G000231_Neeland_",tissue,".Bcell_population.subclusters.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_bcells, file = out2)
}Other Clusters (excluding subclusters)
idx <- which(Idents(merged_obj) %in% c("T cells for reclustering", "B cells for reclustering"))
paed_other <- merged_obj[,-idx]
paed_otherAn object of class Seurat
17973 features across 57193 samples within 1 assay
Active assay: RNA (17973 features, 2000 variable features)
3 layers present: data, counts, scale.data
4 dimensional reductions calculated: pca, umap.unintegrated, harmony, umap.harmonySave subclustered SEU object ( All other cells)
paed_other$cell_labels_v2 <-paed_other$cell_labels
out2 <- here("output",
"RDS", "AllBatches_Annotated_Subclustering_SEUs_v2", tissue,
paste0("G000231_Neeland_",tissue,".all_other.subclusters.SEU.rds"))
if (!file.exists(out2)) {
saveRDS(paed_other, file = out2)
}Merge seurat objects of subclusters
files <- list.files(here("output",
"RDS", "AllBatches_Annotated_Subclustering_SEUs_v2", tissue),
full.names = TRUE)
seuLst <- lapply(files, function(f) readRDS(f))
seu <- merge(seuLst[[1]],
y = c(seuLst[[2]],
seuLst[[3]]))
seuAn object of class Seurat
17973 features across 93547 samples within 1 assay
Active assay: RNA (17973 features, 2000 variable features)
9 layers present: data.1, data.2, data.3, counts.1, scale.data.1, counts.2, scale.data.2, counts.3, scale.data.3levels(seu$cell_labels_v2)[levels(seu$cell_labels_v2) == "ciliated cells"] <- "ciliated epithelial cells"
levels(Idents(seu))[levels(Idents(seu)) == "ciliated cells"] <- "ciliated epithelial cells"
seu$cell_labels_v2 <- Idents(seu)merged <- seu %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()Normalizing layer: counts.1Normalizing layer: counts.2Normalizing layer: counts.3Finding variable features for layer counts.1Finding variable features for layer counts.2Finding variable features for layer counts.3Centering and scaling data matrixPC_ 1
Positive: CORO1A, LAPTM5, LCP1, CD53, SRGN, CXCR4, COTL1, LCP2, ITGB2, IL32
CCL5, TNFRSF1B, SPN, ZEB2, SERPINB9, LTB, CD7, FLNA, EMP3, IL2RB
VIM, SPI1, IRF8, APOBR, CD3E, ALOX5AP, CD2, METRNL, NKG7, ARL4C
Negative: C9orf24, PIFO, ROPN1L, SNTN, RSPH4A, EFCAB1, CAPSL, WDR66, LDLRAD1, DRC3
FAM183A, C11orf88, MORN2, SPA17, CCDC170, C1orf194, SAXO2, DNAH9, CFAP126, CCDC65
C9orf135, FAM81B, ARMC3, WDR49, ERICH3, CC2D2A, FAM216B, RSPH9, NPHP1, AKAP14
PC_ 2
Positive: CORO1A, LAPTM5, LCP1, CD53, VIM, CCL5, IL2RB, COTL1, CD3E, CD2
LTB, SPN, CD8A, CXCR4, NKG7, CD7, IL32, SRGN, ALOX5AP, CD79A
MS4A1, PRF1, GZMA, IGHM, KLHL6, TRBC1, LCP2, CD22, CD69, CCR5
Negative: S100P, WFDC2, LYPD2, AQP3, SDC1, XBP1, GLUL, PI3, CKAP4, GSN
IDO1, SAT1, BPIFA1, PTGES, ALDH3A1, LMNA, SGK1, RARRES1, NECTIN2, SLURP2
GPRC5A, CEBPD, TMEM54, ALPL, SFN, GSTP1, STEAP4, VEGFA, IRS2, S100A9
PC_ 3
Positive: SYNE2, SDC1, PRDX2, CCL5, CD3E, CD2, HIST1H1C, CD8A, IL32, ADGRG1
AQP3, LDHB, MKI67, IL2RB, TYMS, CD7, HIST1H1B, HELLS, KIFC1, TOP2A
TK1, ALDH3A1, FOXM1, ZWINT, GZMA, TRBC1, BIRC5, RRM2, LYPD2, TPX2
Negative: LILRB2, FCER1G, CSF1R, CD14, TYROBP, EMILIN2, CD163, AIF1, LILRA5, SERPINA1
SLC8A1, FPR1, ADGRE2, MEFV, CSF3R, CD68, MS4A6A, HCK, MAFB, STAB1
TGFBI, SPI1, CD300E, PLAUR, KCTD12, TMEM176B, VASH1, IL4I1, APOBEC3A, IFI30
PC_ 4
Positive: S100P, WFDC2, LYPD2, FCGBP, PI3, BPIFA1, XBP1, STEAP4, RARRES1, TFF3
SLC9A3, SPNS2, SLURP2, ALPL, AQP3, ALDH3A1, HID1, DUOXA2, MUC2, GPRC5A
ADGRG1, PTGES, SAT1, IDO1, IRS2, TRIM31, TFF1, VNN3, FA2H, RRBP1
Negative: KIFC1, MKI67, TOP2A, TYMS, AURKB, RRM2, BIRC5, HIST1H1B, NUSAP1, FOXM1
TK1, TPX2, CDK1, HJURP, MYBL2, STMN1, CCNB2, ZWINT, KIF2C, ASF1B
SPC24, UHRF1, NCAPG, CDC20, DLGAP5, KIF23, PLK1, GTSE1, CDCA8, BUB1
PC_ 5
Positive: CD79A, MS4A1, CD22, PAX5, WDFY4, IGKC, IRF8, IGHM, IGHD, BCL11A
POU2AF1, SPIB, FCRL5, BLK, FCRLA, BLNK, TCL1A, TNFRSF13B, MPEG1, CXCR5
IGHG1, BTK, PLCG2, CR1, CYBB, LRMP, CD83, LTB, CTSH, CR2
Negative: CCL5, IL32, CD7, NKG7, CD8A, CD3E, CD2, IL2RB, PRF1, GZMA
LCP2, SPN, LAG3, TRBC1, KLRD1, CST7, GNLY, CTSW, SRGN, GZMB
CCR5, TIGIT, CMTM3, CSF1, JAML, KLRC1, FCRL6, GIMAP4, GBP5, HOPX merged <- RunUMAP(merged, dims = 1:30, reduction = "pca", reduction.name = "umap.merged")21:53:16 UMAP embedding parameters a = 0.9922 b = 1.112Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'21:53:16 Read 93547 rows and found 30 numeric columns21:53:16 Using Annoy for neighbor search, n_neighbors = 30Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'21:53:16 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
21:53:21 Writing NN index file to temp file /var/folders/q8/kw1r78g12qn793xm7g0zvk94x2bh70/T//RtmpPbMoxm/file15b41519a9bb4
21:53:21 Searching Annoy index using 1 thread, search_k = 3000
21:53:39 Annoy recall = 100%
21:53:40 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
21:53:41 Initializing from normalized Laplacian + noise (using RSpectra)
21:53:59 Commencing optimization for 200 epochs, with 4086218 positive edges
21:54:23 Optimization finishedp4 <- DimPlot(merged, reduction = "umap.merged", group.by = "cell_labels_v2",raster = FALSE, repel = TRUE, label = TRUE, label.size = 4.5) + ggtitle(paste0(tissue, ": UMAP with annotations")) + NoLegend()
p4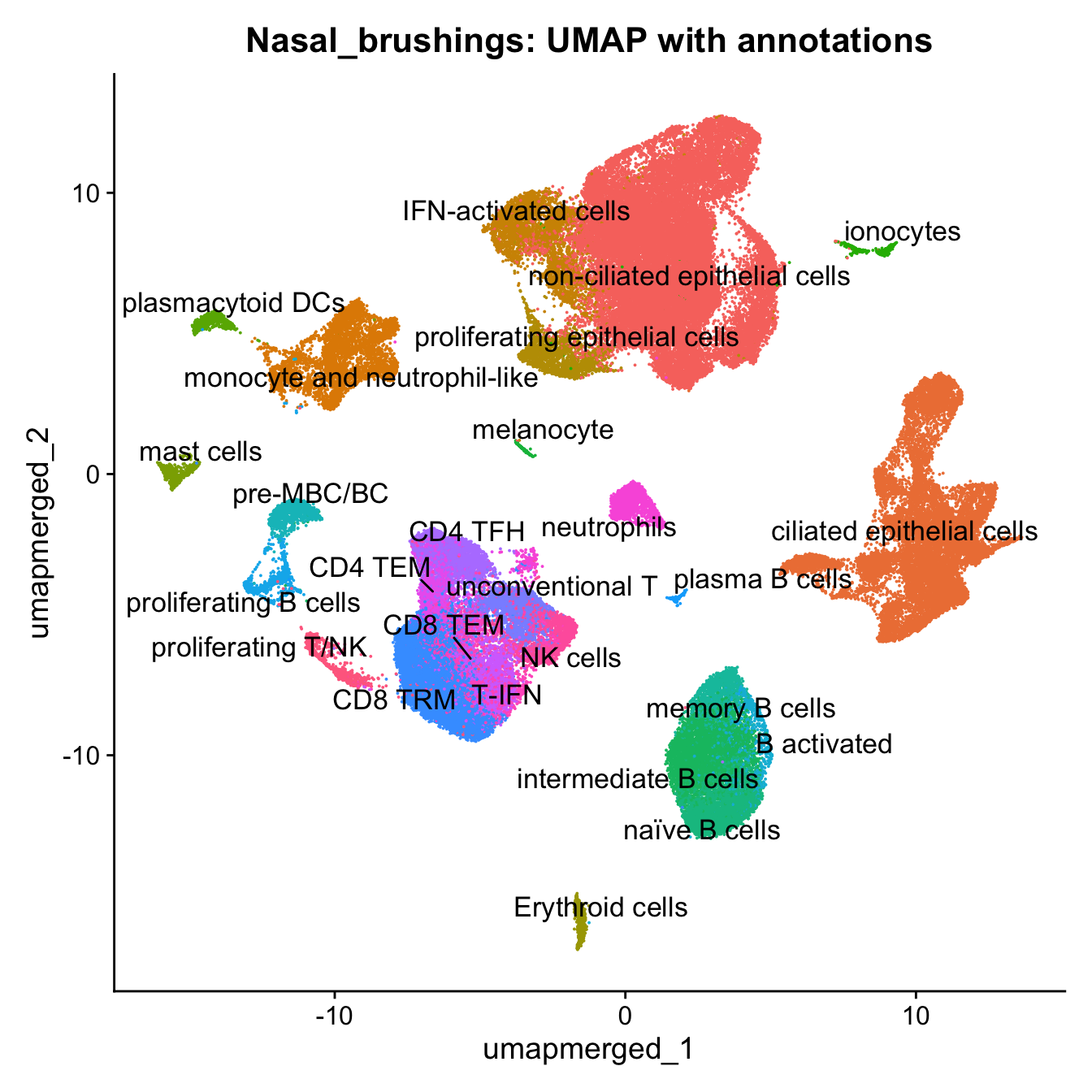
Save Final SEU object (All cells)
out3 <- here("output",
"RDS", "AllBatches_Final_Clusters_SEUs_v2",
paste0("G000231_Neeland_",tissue,".final_clusters.SEU.rds"))
if (!file.exists(out3)) {
saveRDS(merged, file = out3)
}Session Info
sessioninfo::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.3.2 (2023-10-31)
os macOS 15.2
system aarch64, darwin20
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2025-01-15
pandoc 3.1.1 @ /Users/dixitgunjan/Desktop/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
AnnotationDbi * 1.64.1 2023-11-02 [1] Bioconductor
backports 1.4.1 2021-12-13 [1] CRAN (R 4.3.0)
beeswarm 0.4.0 2021-06-01 [1] CRAN (R 4.3.0)
Biobase * 2.62.0 2023-10-26 [1] Bioconductor
BiocGenerics * 0.48.1 2023-11-02 [1] Bioconductor
BiocManager 1.30.22 2023-08-08 [1] CRAN (R 4.3.0)
BiocStyle * 2.30.0 2023-10-26 [1] Bioconductor
Biostrings 2.70.2 2024-01-30 [1] Bioconductor 3.18 (R 4.3.2)
bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
bslib 0.6.1 2023-11-28 [1] CRAN (R 4.3.1)
cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
callr 3.7.5 2024-02-19 [1] CRAN (R 4.3.1)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.3.0)
checkmate 2.3.1 2023-12-04 [1] CRAN (R 4.3.1)
cli 3.6.2 2023-12-11 [1] CRAN (R 4.3.1)
cluster 2.1.6 2023-12-01 [1] CRAN (R 4.3.1)
clustree * 0.5.1 2023-11-05 [1] CRAN (R 4.3.1)
codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.2)
colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
cowplot 1.1.3 2024-01-22 [1] CRAN (R 4.3.1)
crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
data.table * 1.15.0 2024-01-30 [1] CRAN (R 4.3.1)
DBI 1.2.2 2024-02-16 [1] CRAN (R 4.3.1)
DelayedArray 0.28.0 2023-11-06 [1] Bioconductor
deldir 2.0-2 2023-11-23 [1] CRAN (R 4.3.1)
digest 0.6.34 2024-01-11 [1] CRAN (R 4.3.1)
dotCall64 1.1-1 2023-11-28 [1] CRAN (R 4.3.1)
dplyr * 1.1.4 2023-11-17 [1] CRAN (R 4.3.1)
edgeR * 4.0.16 2024-02-20 [1] Bioconductor 3.18 (R 4.3.2)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
evaluate 0.23 2023-11-01 [1] CRAN (R 4.3.1)
fansi 1.0.6 2023-12-08 [1] CRAN (R 4.3.1)
farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
fastDummies 1.7.3 2023-07-06 [1] CRAN (R 4.3.0)
fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
fitdistrplus 1.1-11 2023-04-25 [1] CRAN (R 4.3.0)
forcats * 1.0.0 2023-01-29 [1] CRAN (R 4.3.0)
fs 1.6.3 2023-07-20 [1] CRAN (R 4.3.0)
future 1.33.1 2023-12-22 [1] CRAN (R 4.3.1)
future.apply 1.11.1 2023-12-21 [1] CRAN (R 4.3.1)
generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
GenomeInfoDb 1.38.6 2024-02-10 [1] Bioconductor 3.18 (R 4.3.2)
GenomeInfoDbData 1.2.11 2024-02-27 [1] Bioconductor
GenomicRanges 1.54.1 2023-10-30 [1] Bioconductor
getPass 0.2-4 2023-12-10 [1] CRAN (R 4.3.1)
ggbeeswarm 0.7.2 2023-04-29 [1] CRAN (R 4.3.0)
ggforce 0.4.2 2024-02-19 [1] CRAN (R 4.3.1)
ggplot2 * 3.5.0 2024-02-23 [1] CRAN (R 4.3.1)
ggraph * 2.1.0 2022-10-09 [1] CRAN (R 4.3.0)
ggrastr 1.0.2 2023-06-01 [1] CRAN (R 4.3.0)
ggrepel 0.9.5 2024-01-10 [1] CRAN (R 4.3.1)
ggridges 0.5.6 2024-01-23 [1] CRAN (R 4.3.1)
git2r 0.33.0 2023-11-26 [1] CRAN (R 4.3.1)
globals 0.16.2 2022-11-21 [1] CRAN (R 4.3.0)
glue * 1.7.0 2024-01-09 [1] CRAN (R 4.3.1)
goftest 1.2-3 2021-10-07 [1] CRAN (R 4.3.0)
graphlayouts 1.1.0 2024-01-19 [1] CRAN (R 4.3.1)
gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
gtable 0.3.4 2023-08-21 [1] CRAN (R 4.3.0)
here * 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
htmltools 0.5.7 2023-11-03 [1] CRAN (R 4.3.1)
htmlwidgets 1.6.4 2023-12-06 [1] CRAN (R 4.3.1)
httpuv 1.6.14 2024-01-26 [1] CRAN (R 4.3.1)
httr 1.4.7 2023-08-15 [1] CRAN (R 4.3.0)
ica 1.0-3 2022-07-08 [1] CRAN (R 4.3.0)
igraph 2.0.2 2024-02-17 [1] CRAN (R 4.3.1)
IRanges * 2.36.0 2023-10-26 [1] Bioconductor
irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.2)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
jsonlite 1.8.8 2023-12-04 [1] CRAN (R 4.3.1)
kableExtra * 1.4.0 2024-01-24 [1] CRAN (R 4.3.1)
KEGGREST 1.42.0 2023-10-26 [1] Bioconductor
KernSmooth 2.23-22 2023-07-10 [1] CRAN (R 4.3.2)
knitr 1.45 2023-10-30 [1] CRAN (R 4.3.1)
labeling 0.4.3 2023-08-29 [1] CRAN (R 4.3.0)
later 1.3.2 2023-12-06 [1] CRAN (R 4.3.1)
lattice 0.22-5 2023-10-24 [1] CRAN (R 4.3.1)
lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
leiden 0.4.3.1 2023-11-17 [1] CRAN (R 4.3.1)
lifecycle 1.0.4 2023-11-07 [1] CRAN (R 4.3.1)
limma * 3.58.1 2023-11-02 [1] Bioconductor
listenv 0.9.1 2024-01-29 [1] CRAN (R 4.3.1)
lmtest 0.9-40 2022-03-21 [1] CRAN (R 4.3.0)
locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
lubridate * 1.9.3 2023-09-27 [1] CRAN (R 4.3.1)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
MASS 7.3-60.0.1 2024-01-13 [1] CRAN (R 4.3.1)
Matrix 1.6-5 2024-01-11 [1] CRAN (R 4.3.1)
MatrixGenerics 1.14.0 2023-10-26 [1] Bioconductor
matrixStats 1.2.0 2023-12-11 [1] CRAN (R 4.3.1)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.3.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
nlme 3.1-164 2023-11-27 [1] CRAN (R 4.3.1)
org.Hs.eg.db * 3.18.0 2024-02-27 [1] Bioconductor
paletteer 1.6.0 2024-01-21 [1] CRAN (R 4.3.1)
parallelly 1.37.0 2024-02-14 [1] CRAN (R 4.3.1)
patchwork * 1.2.0 2024-01-08 [1] CRAN (R 4.3.1)
pbapply 1.7-2 2023-06-27 [1] CRAN (R 4.3.0)
pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
plotly 4.10.4 2024-01-13 [1] CRAN (R 4.3.1)
plyr 1.8.9 2023-10-02 [1] CRAN (R 4.3.1)
png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
polyclip 1.10-6 2023-09-27 [1] CRAN (R 4.3.1)
presto 1.0.0 2024-02-27 [1] Github (immunogenomics/presto@31dc97f)
prismatic 1.1.1 2022-08-15 [1] CRAN (R 4.3.0)
processx 3.8.3 2023-12-10 [1] CRAN (R 4.3.1)
progressr 0.14.0 2023-08-10 [1] CRAN (R 4.3.0)
promises 1.2.1 2023-08-10 [1] CRAN (R 4.3.0)
ps 1.7.6 2024-01-18 [1] CRAN (R 4.3.1)
purrr * 1.0.2 2023-08-10 [1] CRAN (R 4.3.0)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
RANN 2.6.1 2019-01-08 [1] CRAN (R 4.3.0)
RColorBrewer * 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
Rcpp 1.0.12 2024-01-09 [1] CRAN (R 4.3.1)
RcppAnnoy 0.0.22 2024-01-23 [1] CRAN (R 4.3.1)
RcppHNSW 0.6.0 2024-02-04 [1] CRAN (R 4.3.1)
RCurl 1.98-1.14 2024-01-09 [1] CRAN (R 4.3.1)
readr * 2.1.5 2024-01-10 [1] CRAN (R 4.3.1)
readxl * 1.4.3 2023-07-06 [1] CRAN (R 4.3.0)
rematch2 2.1.2 2020-05-01 [1] CRAN (R 4.3.0)
reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
reticulate 1.35.0 2024-01-31 [1] CRAN (R 4.3.1)
rlang 1.1.3 2024-01-10 [1] CRAN (R 4.3.1)
rmarkdown 2.25 2023-09-18 [1] CRAN (R 4.3.1)
ROCR 1.0-11 2020-05-02 [1] CRAN (R 4.3.0)
rprojroot 2.0.4 2023-11-05 [1] CRAN (R 4.3.1)
RSpectra 0.16-1 2022-04-24 [1] CRAN (R 4.3.0)
RSQLite 2.3.5 2024-01-21 [1] CRAN (R 4.3.1)
rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
Rtsne 0.17 2023-12-07 [1] CRAN (R 4.3.1)
S4Arrays 1.2.0 2023-10-26 [1] Bioconductor
S4Vectors * 0.40.2 2023-11-25 [1] Bioconductor 3.18 (R 4.3.2)
sass 0.4.8 2023-12-06 [1] CRAN (R 4.3.1)
scales 1.3.0 2023-11-28 [1] CRAN (R 4.3.1)
scattermore 1.2 2023-06-12 [1] CRAN (R 4.3.0)
sctransform 0.4.1 2023-10-19 [1] CRAN (R 4.3.1)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
Seurat * 5.0.1.9009 2024-02-28 [1] Github (satijalab/seurat@6a3ef5e)
SeuratObject * 5.0.1 2023-11-17 [1] CRAN (R 4.3.1)
shiny 1.8.0 2023-11-17 [1] CRAN (R 4.3.1)
SingleCellExperiment 1.24.0 2023-11-06 [1] Bioconductor
sp * 2.1-3 2024-01-30 [1] CRAN (R 4.3.1)
spam 2.10-0 2023-10-23 [1] CRAN (R 4.3.1)
SparseArray 1.2.4 2024-02-10 [1] Bioconductor 3.18 (R 4.3.2)
spatstat.data 3.0-4 2024-01-15 [1] CRAN (R 4.3.1)
spatstat.explore 3.2-6 2024-02-01 [1] CRAN (R 4.3.1)
spatstat.geom 3.2-8 2024-01-26 [1] CRAN (R 4.3.1)
spatstat.random 3.2-2 2023-11-29 [1] CRAN (R 4.3.1)
spatstat.sparse 3.0-3 2023-10-24 [1] CRAN (R 4.3.1)
spatstat.utils 3.0-4 2023-10-24 [1] CRAN (R 4.3.1)
speckle * 1.2.0 2023-10-26 [1] Bioconductor
statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
stringi 1.8.3 2023-12-11 [1] CRAN (R 4.3.1)
stringr * 1.5.1 2023-11-14 [1] CRAN (R 4.3.1)
SummarizedExperiment 1.32.0 2023-11-06 [1] Bioconductor
survival 3.5-8 2024-02-14 [1] CRAN (R 4.3.1)
svglite 2.1.3 2023-12-08 [1] CRAN (R 4.3.1)
systemfonts 1.0.5 2023-10-09 [1] CRAN (R 4.3.1)
tensor 1.5 2012-05-05 [1] CRAN (R 4.3.0)
tibble * 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
tidygraph 1.3.1 2024-01-30 [1] CRAN (R 4.3.1)
tidyr * 1.3.1 2024-01-24 [1] CRAN (R 4.3.1)
tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
tidyverse * 2.0.0 2023-02-22 [1] CRAN (R 4.3.0)
timechange 0.3.0 2024-01-18 [1] CRAN (R 4.3.1)
tweenr 2.0.3 2024-02-26 [1] CRAN (R 4.3.1)
tzdb 0.4.0 2023-05-12 [1] CRAN (R 4.3.0)
utf8 1.2.4 2023-10-22 [1] CRAN (R 4.3.1)
uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
vctrs 0.6.5 2023-12-01 [1] CRAN (R 4.3.1)
vipor 0.4.7 2023-12-18 [1] CRAN (R 4.3.1)
viridis 0.6.5 2024-01-29 [1] CRAN (R 4.3.1)
viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
whisker 0.4.1 2022-12-05 [1] CRAN (R 4.3.0)
withr 3.0.0 2024-01-16 [1] CRAN (R 4.3.1)
workflowr * 1.7.1 2023-08-23 [1] CRAN (R 4.3.0)
xfun 0.42 2024-02-08 [1] CRAN (R 4.3.1)
xml2 1.3.6 2023-12-04 [1] CRAN (R 4.3.1)
xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
XVector 0.42.0 2023-10-26 [1] Bioconductor
yaml 2.3.8 2023-12-11 [1] CRAN (R 4.3.1)
zlibbioc 1.48.0 2023-10-26 [1] Bioconductor
zoo 1.8-12 2023-04-13 [1] CRAN (R 4.3.0)
[1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
──────────────────────────────────────────────────────────────────────────────
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.2
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] readxl_1.4.3 org.Hs.eg.db_3.18.0 AnnotationDbi_1.64.1
[4] IRanges_2.36.0 S4Vectors_0.40.2 Biobase_2.62.0
[7] BiocGenerics_0.48.1 speckle_1.2.0 edgeR_4.0.16
[10] limma_3.58.1 patchwork_1.2.0 data.table_1.15.0
[13] RColorBrewer_1.1-3 kableExtra_1.4.0 clustree_0.5.1
[16] ggraph_2.1.0 Seurat_5.0.1.9009 SeuratObject_5.0.1
[19] sp_2.1-3 glue_1.7.0 here_1.0.1
[22] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1
[25] dplyr_1.1.4 purrr_1.0.2 readr_2.1.5
[28] tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.0
[31] tidyverse_2.0.0 BiocStyle_2.30.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.3 matrixStats_1.2.0
[3] spatstat.sparse_3.0-3 bitops_1.0-7
[5] httr_1.4.7 tools_4.3.2
[7] sctransform_0.4.1 backports_1.4.1
[9] utf8_1.2.4 R6_2.5.1
[11] lazyeval_0.2.2 uwot_0.1.16
[13] withr_3.0.0 gridExtra_2.3
[15] progressr_0.14.0 cli_3.6.2
[17] spatstat.explore_3.2-6 fastDummies_1.7.3
[19] prismatic_1.1.1 labeling_0.4.3
[21] sass_0.4.8 spatstat.data_3.0-4
[23] ggridges_0.5.6 pbapply_1.7-2
[25] systemfonts_1.0.5 svglite_2.1.3
[27] sessioninfo_1.2.2 parallelly_1.37.0
[29] rstudioapi_0.15.0 RSQLite_2.3.5
[31] generics_0.1.3 ica_1.0-3
[33] spatstat.random_3.2-2 Matrix_1.6-5
[35] ggbeeswarm_0.7.2 fansi_1.0.6
[37] abind_1.4-5 lifecycle_1.0.4
[39] whisker_0.4.1 yaml_2.3.8
[41] SummarizedExperiment_1.32.0 SparseArray_1.2.4
[43] Rtsne_0.17 paletteer_1.6.0
[45] grid_4.3.2 blob_1.2.4
[47] promises_1.2.1 crayon_1.5.2
[49] miniUI_0.1.1.1 lattice_0.22-5
[51] cowplot_1.1.3 KEGGREST_1.42.0
[53] pillar_1.9.0 knitr_1.45
[55] GenomicRanges_1.54.1 future.apply_1.11.1
[57] codetools_0.2-19 leiden_0.4.3.1
[59] getPass_0.2-4 vctrs_0.6.5
[61] png_0.1-8 spam_2.10-0
[63] cellranger_1.1.0 gtable_0.3.4
[65] rematch2_2.1.2 cachem_1.0.8
[67] xfun_0.42 S4Arrays_1.2.0
[69] mime_0.12 tidygraph_1.3.1
[71] survival_3.5-8 SingleCellExperiment_1.24.0
[73] statmod_1.5.0 ellipsis_0.3.2
[75] fitdistrplus_1.1-11 ROCR_1.0-11
[77] nlme_3.1-164 bit64_4.0.5
[79] RcppAnnoy_0.0.22 GenomeInfoDb_1.38.6
[81] rprojroot_2.0.4 bslib_0.6.1
[83] irlba_2.3.5.1 vipor_0.4.7
[85] KernSmooth_2.23-22 colorspace_2.1-0
[87] DBI_1.2.2 ggrastr_1.0.2
[89] tidyselect_1.2.0 processx_3.8.3
[91] bit_4.0.5 compiler_4.3.2
[93] git2r_0.33.0 xml2_1.3.6
[95] DelayedArray_0.28.0 plotly_4.10.4
[97] checkmate_2.3.1 scales_1.3.0
[99] lmtest_0.9-40 callr_3.7.5
[101] digest_0.6.34 goftest_1.2-3
[103] spatstat.utils_3.0-4 presto_1.0.0
[105] rmarkdown_2.25 XVector_0.42.0
[107] htmltools_0.5.7 pkgconfig_2.0.3
[109] MatrixGenerics_1.14.0 highr_0.10
[111] fastmap_1.1.1 rlang_1.1.3
[113] htmlwidgets_1.6.4 shiny_1.8.0
[115] farver_2.1.1 jquerylib_0.1.4
[117] zoo_1.8-12 jsonlite_1.8.8
[119] RCurl_1.98-1.14 magrittr_2.0.3
[121] GenomeInfoDbData_1.2.11 dotCall64_1.1-1
[123] munsell_0.5.0 Rcpp_1.0.12
[125] viridis_0.6.5 reticulate_1.35.0
[127] stringi_1.8.3 zlibbioc_1.48.0
[129] MASS_7.3-60.0.1 plyr_1.8.9
[131] parallel_4.3.2 listenv_0.9.1
[133] ggrepel_0.9.5 deldir_2.0-2
[135] Biostrings_2.70.2 graphlayouts_1.1.0
[137] splines_4.3.2 tensor_1.5
[139] hms_1.1.3 locfit_1.5-9.8
[141] ps_1.7.6 igraph_2.0.2
[143] spatstat.geom_3.2-8 RcppHNSW_0.6.0
[145] reshape2_1.4.4 evaluate_0.23
[147] BiocManager_1.30.22 tzdb_0.4.0
[149] tweenr_2.0.3 httpuv_1.6.14
[151] RANN_2.6.1 polyclip_1.10-6
[153] future_1.33.1 scattermore_1.2
[155] ggforce_0.4.2 xtable_1.8-4
[157] RSpectra_0.16-1 later_1.3.2
[159] viridisLite_0.4.2 beeswarm_0.4.0
[161] memoise_2.0.1 cluster_2.1.6
[163] timechange_0.3.0 globals_0.16.2