Subclustering: BAL
Unsupervised Clustering of Broad cell labels
Gunjan Dixit
October 11, 2024
Last updated: 2024-10-11
Checks: 6 1
Knit directory: paed-airway-allTissues/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown file has unstaged changes. To know which version of
the R Markdown file created these results, you’ll want to first commit
it to the Git repo. If you’re still working on the analysis, you can
ignore this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230811) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 8f254aa. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: data/.DS_Store
Ignored: data/Cell_labels_Mel_v3/
Ignored: data/RDS/
Ignored: output/.DS_Store
Ignored: output/CSV/.DS_Store
Ignored: output/G000231_Neeland_batch1/
Ignored: output/G000231_Neeland_batch2_1/
Ignored: output/G000231_Neeland_batch2_2/
Ignored: output/G000231_Neeland_batch3/
Ignored: output/G000231_Neeland_batch4/
Ignored: output/G000231_Neeland_batch5/
Ignored: output/G000231_Neeland_batch9_1/
Ignored: output/RDS/
Ignored: output/plots/
Untracked files:
Untracked: Adenoids_Bcell_subset_proportions_Age.pdf
Untracked: Adenoids_Tcell_subset_proportions_Age.pdf
Untracked: Adenoids_cell_type_proportions_Age.pdf
Untracked: Age_proportions_Adenoids.pdf
Untracked: Age_proportions_Bronchial_brushings.pdf
Untracked: Age_proportions_Nasal_brushings.pdf
Untracked: Age_proportions_Tonsils.pdf
Untracked: BAL_Tcell_propeller.xlsx
Untracked: BAL_propeller.xlsx
Untracked: BB_Tcell_propeller.xlsx
Untracked: BB_propeller.xlsx
Untracked: NB_Tcell_propeller.xlsx
Untracked: NB_propeller.csv
Untracked: NB_propeller.pdf
Untracked: NB_propeller.xlsx
Untracked: Tonsils_cell_type_proportions.jpg
Untracked: Tonsils_cell_type_proportions.pdf
Untracked: Tonsils_cell_type_proportions.png
Untracked: Tonsils_cell_type_proportions_Age.pdf
Untracked: analysis/03_Batch_Integration.Rmd
Untracked: analysis/Age_proportions.Rmd
Untracked: analysis/Age_proportions_AllBatches.Rmd
Untracked: analysis/Batch_Integration_&_Downstream_analysis.Rmd
Untracked: analysis/Batch_correction_&_Downstream.Rmd
Untracked: analysis/Cell_cycle_regression.Rmd
Untracked: analysis/Master_metadata.Rmd
Untracked: analysis/Preprocessing_Batch1_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch2_Tonsils.Rmd
Untracked: analysis/Preprocessing_Batch3_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch4_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch5_Nasal_brushings.Rmd
Untracked: analysis/Preprocessing_Batch6_BAL.Rmd
Untracked: analysis/Preprocessing_Batch7_Bronchial_brushings.Rmd
Untracked: analysis/Preprocessing_Batch8_Adenoids.Rmd
Untracked: analysis/Preprocessing_Batch9_Tonsils.Rmd
Untracked: analysis/TonsilsVsAdenoids.Rmd
Untracked: analysis/cell_cycle_regression.R
Untracked: analysis/test.Rmd
Untracked: analysis/testing_age_all.Rmd
Untracked: cell_proportions_overview.png
Untracked: cell_type_proportions.pdf
Untracked: cell_type_proportions_enhanced.pdf
Untracked: cell_type_proportions_individual.pdf
Untracked: color_palette.rds
Untracked: color_palette_v2_level2.rds
Untracked: combined_metadata.rds
Untracked: data/Cell_labels_Mel/
Untracked: data/Cell_labels_Mel_v2/
Untracked: data/Cell_labels_modified_Gunjan/
Untracked: data/Hs.c2.cp.reactome.v7.1.entrez.rds
Untracked: data/Raw_feature_bc_matrix/
Untracked: data/celltypes_Mel_GD_v3.xlsx
Untracked: data/celltypes_Mel_GD_v4_no_dups.xlsx
Untracked: data/celltypes_Mel_modified.xlsx
Untracked: data/celltypes_Mel_v2.csv
Untracked: data/celltypes_Mel_v2.xlsx
Untracked: data/celltypes_Mel_v2_MN.xlsx
Untracked: data/celltypes_for_mel_MN.xlsx
Untracked: data/earlyAIR_sample_sheets_combined.xlsx
Untracked: output/CSV/All_tissues.propeller.xlsx
Untracked: output/CSV/Bronchial_brushings/
Untracked: output/CSV/Bronchial_brushings_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/
Untracked: output/CSV/G000231_Neeland_Adenoids.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Bronchial_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Nasal_brushings.propeller.xlsx
Untracked: output/CSV/G000231_Neeland_Tonsils.propeller.xlsx
Untracked: output/CSV/Nasal_brushings/
Unstaged changes:
Deleted: 02_QC_exploratoryPlots.Rmd
Deleted: 02_QC_exploratoryPlots.html
Modified: analysis/00_AllBatches_overview.Rmd
Modified: analysis/01_QC_emptyDrops.Rmd
Modified: analysis/02_QC_exploratoryPlots.Rmd
Modified: analysis/Adenoids.Rmd
Modified: analysis/Age_modeling.Rmd
Modified: analysis/AllBatches_QCExploratory.Rmd
Modified: analysis/BAL.Rmd
Modified: analysis/Bronchial_brushings.Rmd
Modified: analysis/Nasal_brushings.Rmd
Modified: analysis/Subclustering_BAL.Rmd
Modified: analysis/Subclustering_Nasal_brushings.Rmd
Modified: analysis/Tonsils.Rmd
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/REACTOME-cluster-limma-c9.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c0.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c1.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c10.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c11.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c12.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c13.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c14.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c15.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c16.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c17.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c2.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c3.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c4.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c5.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c6.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c7.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c8.csv
Modified: output/CSV/BAL_Marker_gene_clusters.limmaTrendRNA_snn_res.0.4/up-cluster-limma-c9.csv
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/Subclustering_BAL.Rmd) and
HTML (docs/Subclustering_BAL.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 07af966 | Gunjan Dixit | 2024-09-25 | Modified index |
| Rmd | 3b5ab22 | Gunjan Dixit | 2024-09-25 | Separated Subclustering Rmd |
Introduction
Load libraries
suppressPackageStartupMessages({
library(BiocStyle)
library(tidyverse)
library(here)
library(glue)
library(dplyr)
library(Seurat)
library(clustree)
library(kableExtra)
library(RColorBrewer)
library(data.table)
library(ggplot2)
library(patchwork)
library(limma)
library(edgeR)
library(speckle)
library(AnnotationDbi)
library(org.Hs.eg.db)
library(readxl)
})Load Input data
For Bronchial brushings, we used only Batch4 for the downstream analysis.
tissue <- "BAL"
out1 <- here("output",
"RDS", "AllBatches_Clustering_SEUs",
paste0("G000231_Neeland_",tissue,".Clusters.SEU.rds"))
seu_obj <- readRDS(out1)
seu_objAn object of class Seurat
17529 features across 42312 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, umap, umap.unintegratedReclustering Macro polulation
The marker genes for this reclustering can be found here-
idx <- which(Idents(seu_obj) %in% c("macro-monocyte-derived-or-interstitial", "macro-proliferating", "macro-lipid", "macro-alveolar", "macro-CCL"))
paed_sub <- seu_obj[,idx]
mito_genes <- grep("^MT-", rownames(paed_sub), value = TRUE)
#paed_sub@meta.data$donor <- sub("_\\d+$", "", paed_sub@meta.data$donor_id)
paed_sub <- subset(paed_sub, features = setdiff(rownames(paed_sub), mito_genes))
paed_subAn object of class Seurat
17518 features across 32160 samples within 1 assay
Active assay: RNA (17518 features, 1990 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, umap, umap.unintegratedpaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9661
Number of communities: 8
Elapsed time: 24 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9505
Number of communities: 9
Elapsed time: 24 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9356
Number of communities: 11
Elapsed time: 23 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9215
Number of communities: 15
Elapsed time: 22 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9121
Number of communities: 18
Elapsed time: 21 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9039
Number of communities: 18
Elapsed time: 20 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8958
Number of communities: 18
Elapsed time: 20 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8877
Number of communities: 19
Elapsed time: 19 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8805
Number of communities: 20
Elapsed time: 18 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 32160
Number of edges: 1020809
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8735
Number of communities: 21
Elapsed time: 18 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
clustree(paed_sub, prefix = "RNA_snn_res.")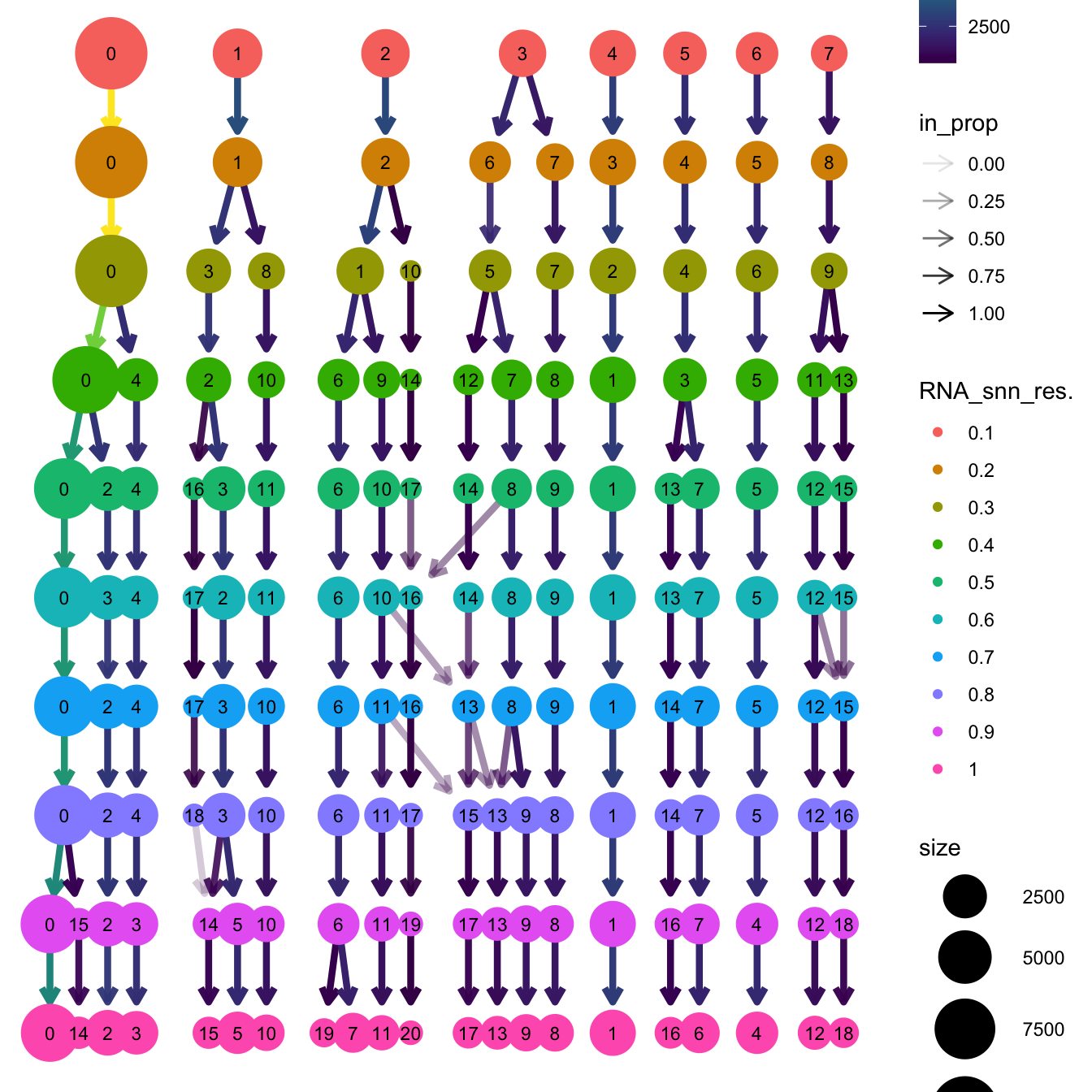
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
opt_res <- "RNA_snn_res.0.7"
n <- nlevels(paed_sub$RNA_snn_res.0.7)
paed_sub$RNA_snn_res.0.7 <- factor(paed_sub$RNA_snn_res.0.7, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
Idents(paed_sub) <- paed_sub$RNA_snn_res.0.7DimPlot(paed_sub, reduction = "umap.new", group.by = "RNA_snn_res.0.7", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )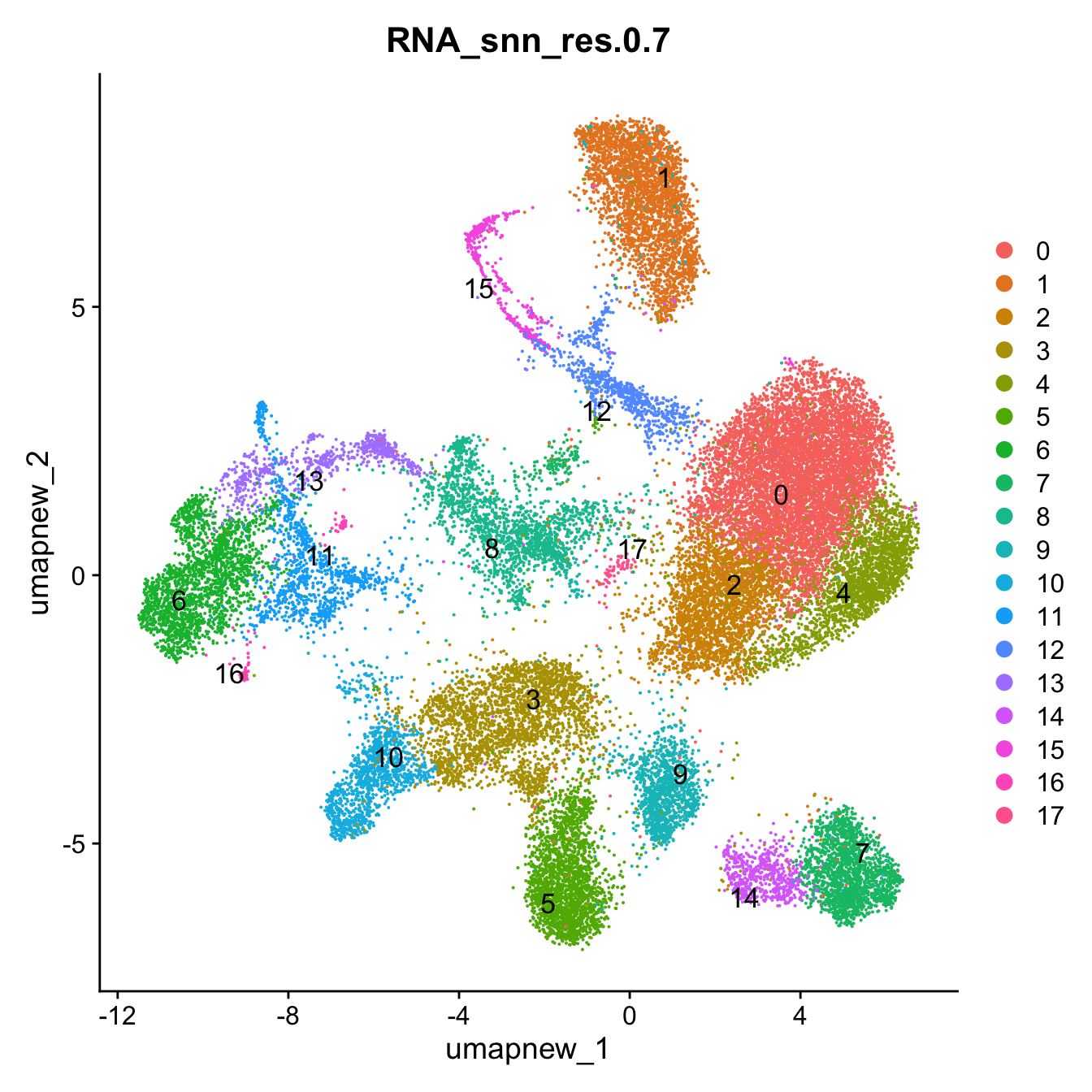
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_finest_level", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )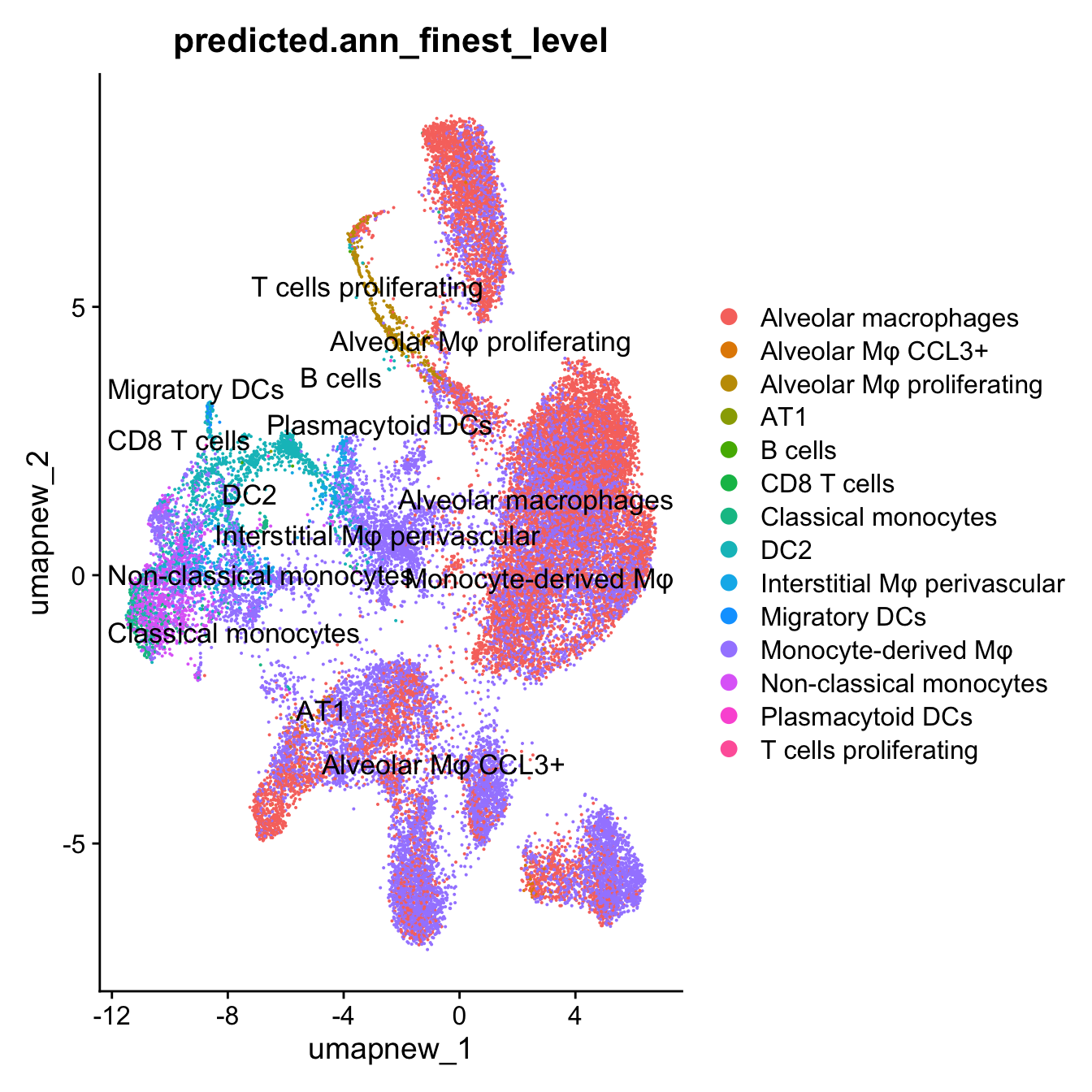
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_level_3", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )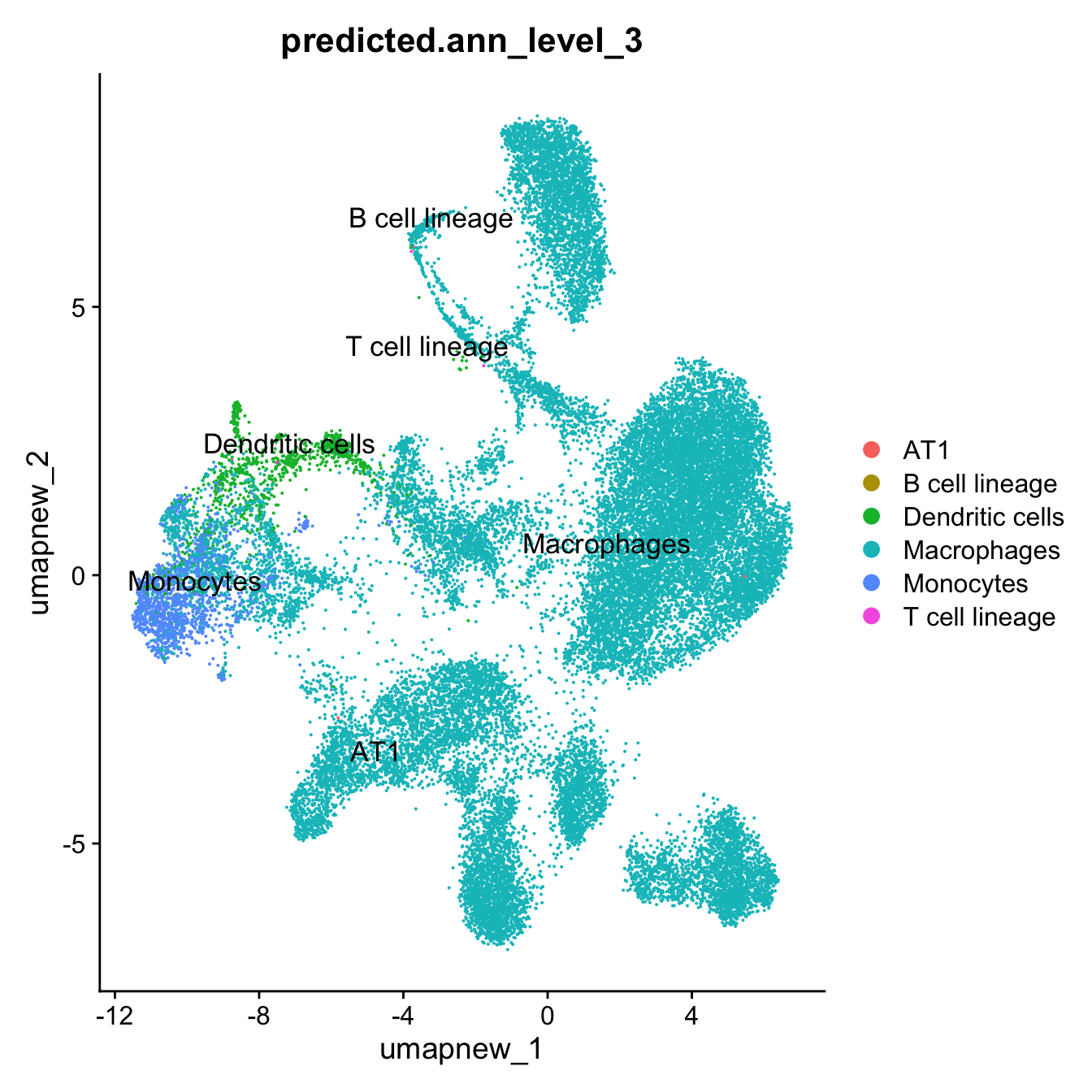
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_level_4", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )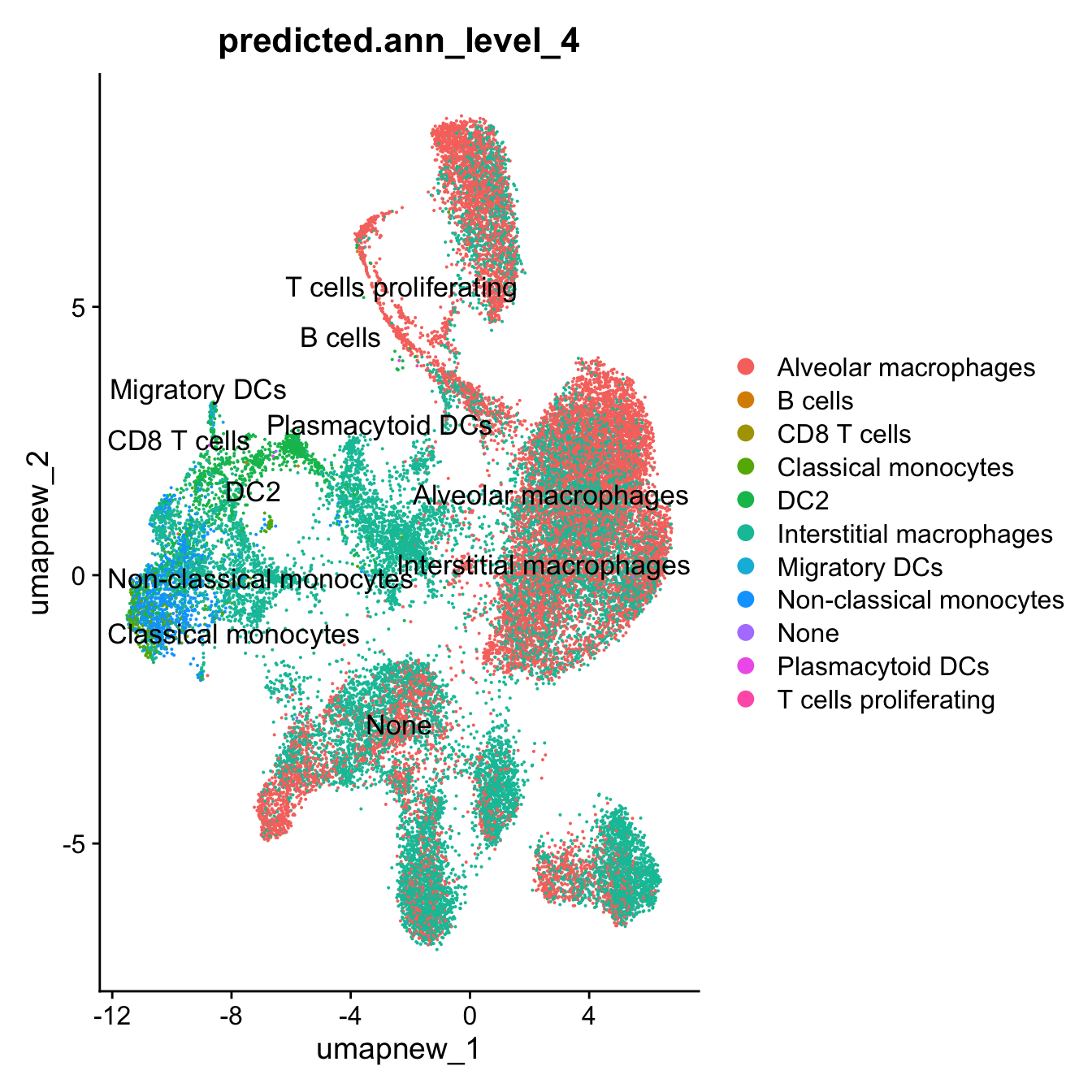
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
DimPlot(paed_sub, reduction = "umap.new", group.by = "predicted.ann_level_5", label = TRUE, label.size = 4.5, repel = TRUE, raster = FALSE )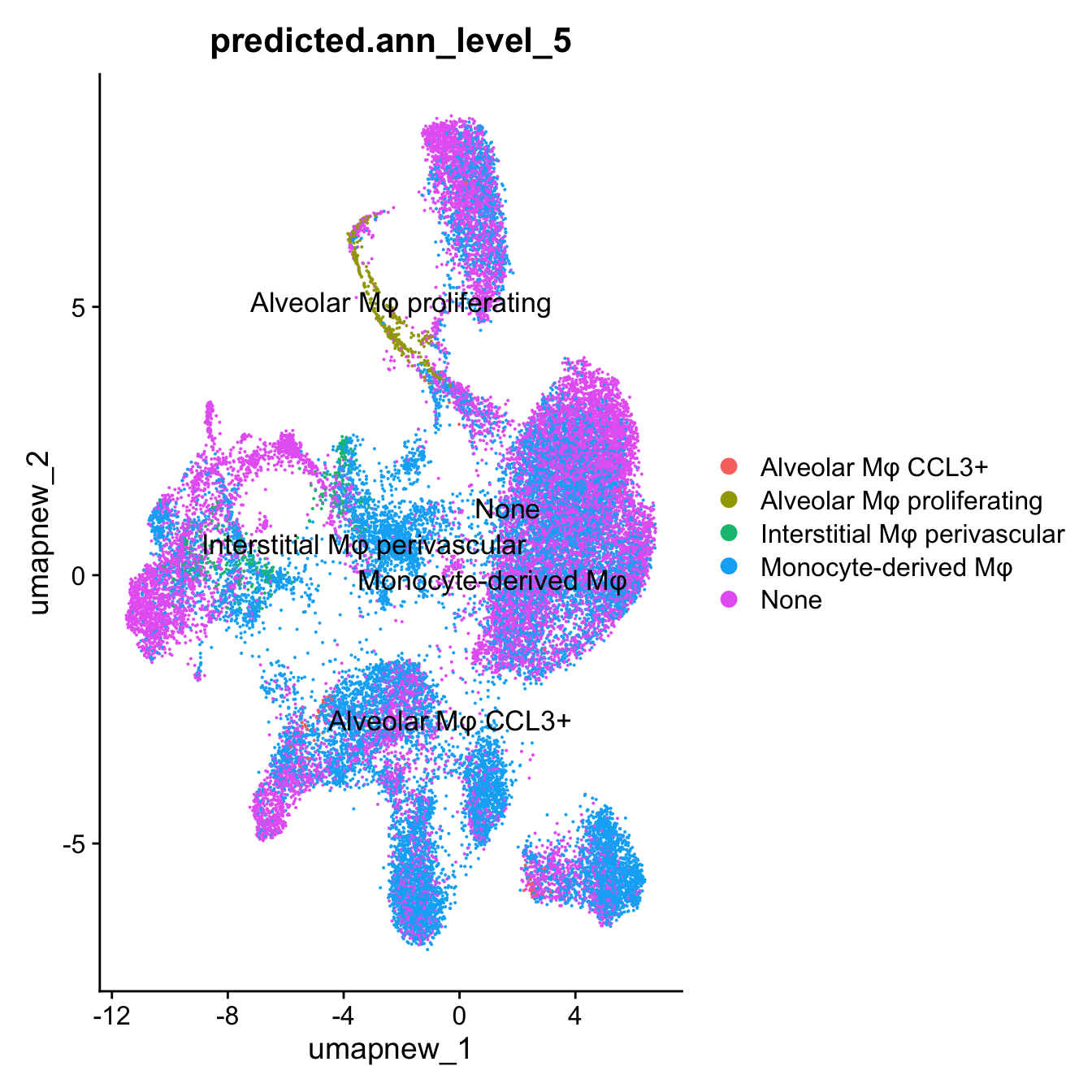
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
paed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8Calculating cluster 9Calculating cluster 10Calculating cluster 11Calculating cluster 12Calculating cluster 13Calculating cluster 14Calculating cluster 15Calculating cluster 16Calculating cluster 17paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 10, wt = avg_log2FC) -> top10
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "THBS1" "DEFB1" "APP" "NR1H3" "SENP3" "PRDX2"
[7] "SERPINB9" "NRP2" "VCAN" "PLA1A" "CXCL2" "SOCS3"
[13] "E2F1" "CLEC10A" "NRP2" "MKI67" "PROK2" "TMSB4X" FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, label = T, ncol = 3)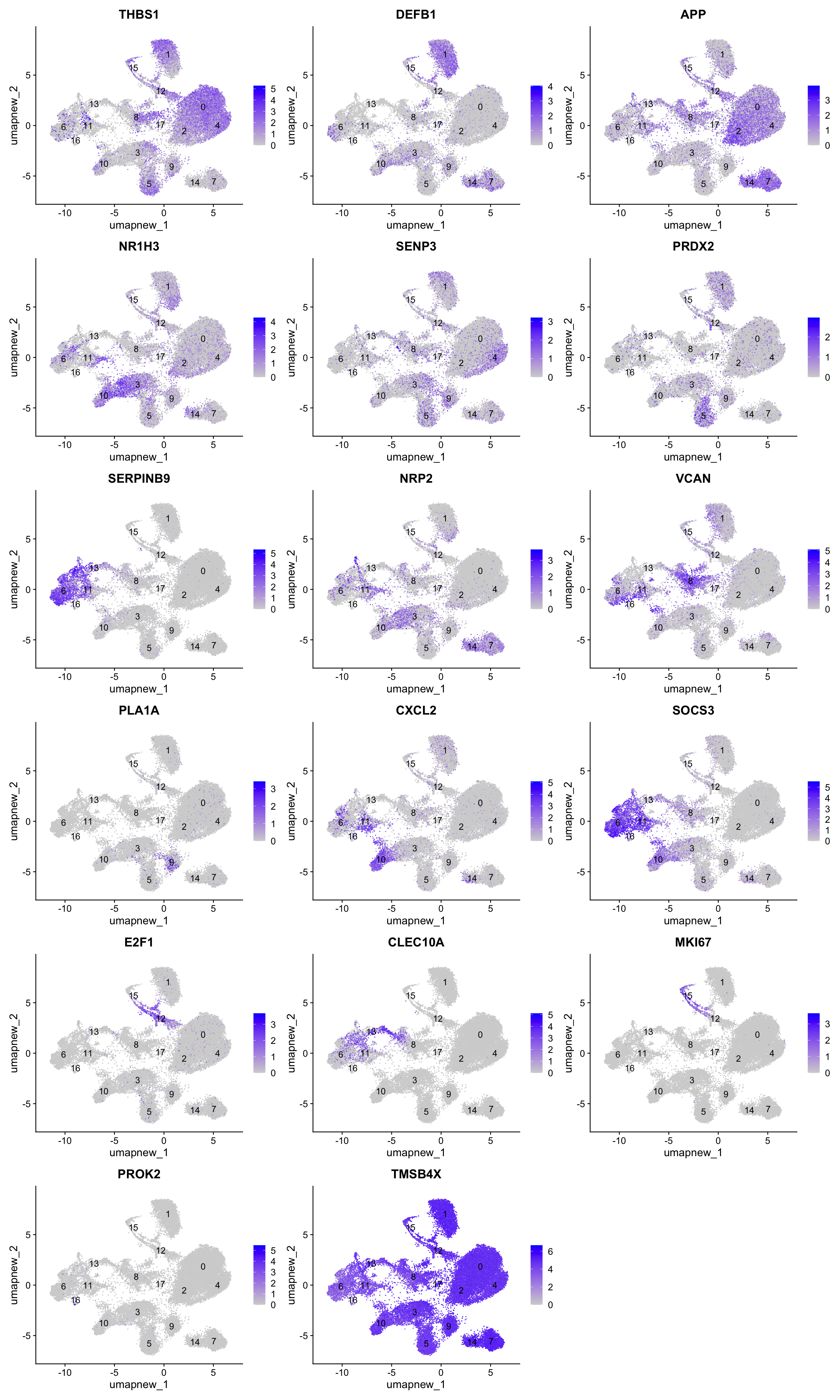
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_macro_population.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
if (!file.exists(file_name)) {
write.csv(cluster_data, file = file_name)
}
}Marker-analysis of Macro population using Limma
paed_sub@meta.data$donor <- sub("_\\d+$", "", paed_sub@meta.data$Sample)
logcounts <- normCounts(DGEList(as.matrix(paed_sub[["RNA"]]$counts)),
log = TRUE, prior.count = 0.5)Warning in asMethod(object): sparse->dense coercion: allocating vector of size
4.2 GiBentrez <- AnnotationDbi::mapIds(org.Hs.eg.db,
keys = rownames(logcounts),
column = c("ENTREZID"),
keytype = "SYMBOL",
multiVals = "first")'select()' returned 1:many mapping between keys and columnslogcounts <- logcounts[!is.na(entrez),]
maxclust <- length(levels(Idents(paed_sub))) - 1
clustgrp <- paste0("c", Idents(paed_sub))
clustgrp <- factor(clustgrp, levels = paste0("c", 0:maxclust))
sample <- paed_sub$donor
design <- model.matrix(~ 0 + clustgrp + sample)
colnames(design)[1:(length(levels(clustgrp)))] <- levels(clustgrp)
head(design) c0 c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11 c12 c13 c14 c15 c16 c17 sampleeAIR035
1 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
2 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
3 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
4 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
5 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
6 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
sampleeAIR036 sampleeAIR045 sampleeAIR046 sampleeAIR054 sampleeAIR057
1 0 0 0 0 0
2 0 0 0 0 0
3 0 0 0 0 0
4 0 0 0 0 0
5 0 0 0 0 0
6 0 0 0 0 0
sampleeAIR059
1 0
2 0
3 0
4 0
5 0
6 0# Create contrast matrix
mycont <- matrix(NA, ncol = length(levels(clustgrp)),
nrow = length(levels(clustgrp)))
rownames(mycont) <- colnames(mycont) <- levels(clustgrp)
diag(mycont) <- 1
mycont[upper.tri(mycont)] <- -1/(length(levels(factor(clustgrp))) - 1)
mycont[lower.tri(mycont)] <- -1/(length(levels(factor(clustgrp))) - 1)
mycont c0 c1 c2 c3 c4 c5
c0 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c1 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c2 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353
c3 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353
c4 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353
c5 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000
c6 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c7 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c8 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c9 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c10 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c11 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c12 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c13 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c14 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c15 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c16 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c17 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c6 c7 c8 c9 c10 c11
c0 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c1 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c2 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c3 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c4 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c5 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c6 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c7 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c8 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353
c9 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353
c10 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353
c11 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000
c12 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c13 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c14 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c15 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c16 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c17 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c12 c13 c14 c15 c16 c17
c0 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c1 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c2 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c3 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c4 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c5 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c6 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c7 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c8 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c9 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c10 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c11 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c12 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c13 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353 -0.05882353
c14 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353 -0.05882353
c15 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353 -0.05882353
c16 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000 -0.05882353
c17 -0.05882353 -0.05882353 -0.05882353 -0.05882353 -0.05882353 1.00000000# Fill out remaining rows with 0s
zero.rows <- matrix(0, ncol = length(levels(clustgrp)),
nrow = (ncol(design) - length(levels(clustgrp))))
fullcont <- rbind(mycont, zero.rows)
rownames(fullcont) <- colnames(design)
fit <- lmFit(logcounts, design)
fit.cont <- contrasts.fit(fit, contrasts = fullcont)
fit.cont <- eBayes(fit.cont, trend = TRUE, robust = TRUE)Warning: 2233 very small variances detected, have been offset away from zerosummary(decideTests(fit.cont)) c0 c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11
Down 2851 1462 2185 1572 1194 1432 8242 1405 2855 1193 2121 5726
NotSig 11300 11435 11419 12200 9311 12279 7103 11554 11675 11326 13173 9060
Up 2960 4214 3507 3339 6606 3400 1766 4152 2581 4592 1817 2325
c12 c13 c14 c15 c16 c17
Down 1095 4218 1065 1136 5477 4738
NotSig 11392 10052 12769 11430 10915 12255
Up 4624 2841 3277 4545 719 118Test relative to a threshold (TREAT)
#tr <- treat(fit.cont, fc=1.1) #10% fold change from documentation
tr <- treat(fit.cont, lfc=0.25)
dt <- decideTests(tr)
summary(dt) c0 c1 c2 c3 c4 c5 c6 c7 c8 c9 c10 c11
Down 104 62 106 91 56 144 1472 84 236 98 119 825
NotSig 16623 16549 16484 16754 16525 16564 15154 16401 16691 16541 16672 15836
Up 384 500 521 266 530 403 485 626 184 472 320 450
c12 c13 c14 c15 c16 c17
Down 84 860 47 137 1492 853
NotSig 16446 15700 16575 16210 15391 16254
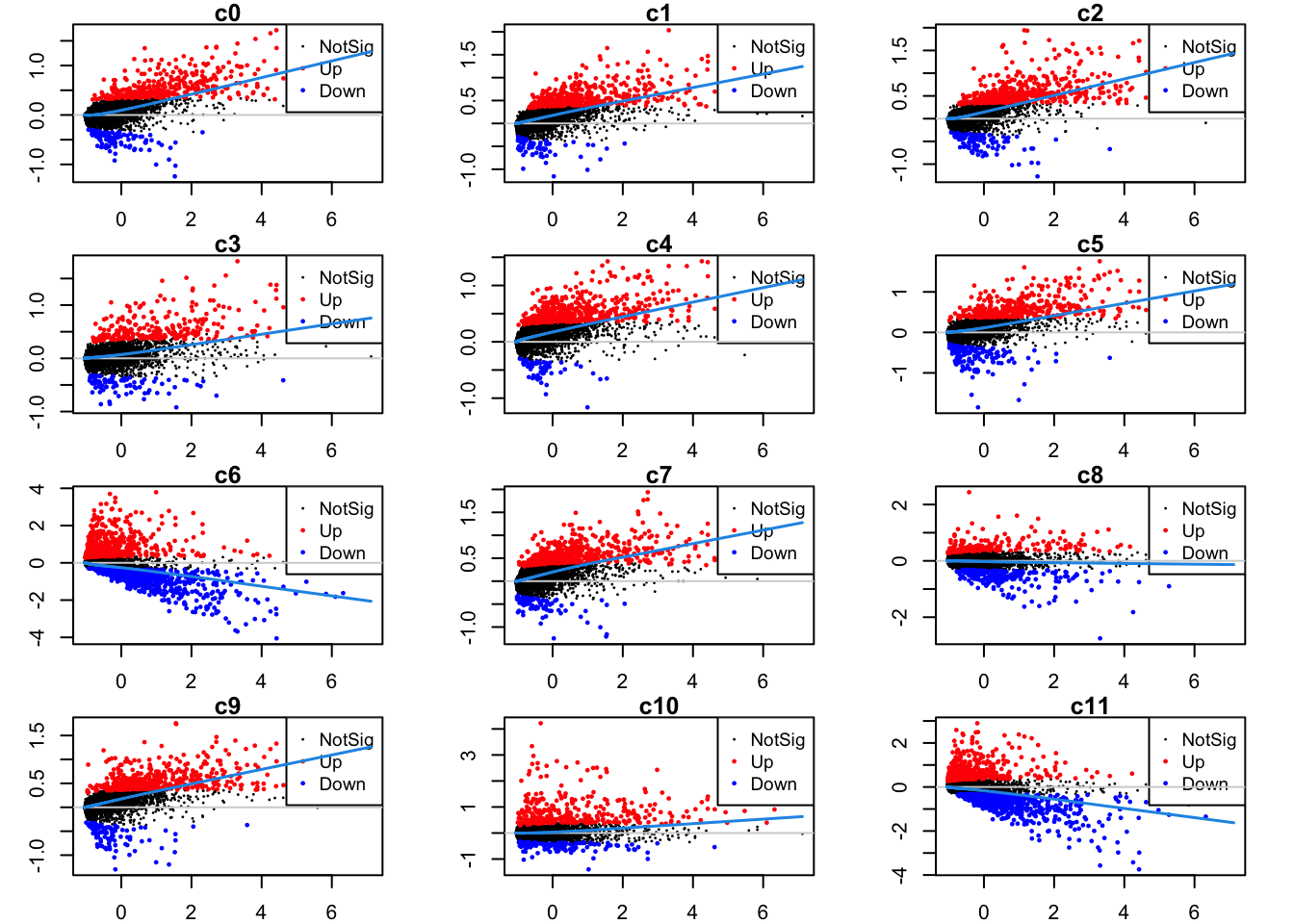
Up 581 551 489 764 228 4Mean-difference plots per cluster
par(mfrow=c(4,3))
par(mar=c(2,3,1,2))
for(i in 1:ncol(mycont)){
plotMD(tr, coef = i, status = dt[,i], hl.cex = 0.5)
abline(h = 0, col = "lightgrey")
lines(lowess(tr$Amean, tr$coefficients[,i]), lwd = 1.5, col = 4)
}
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
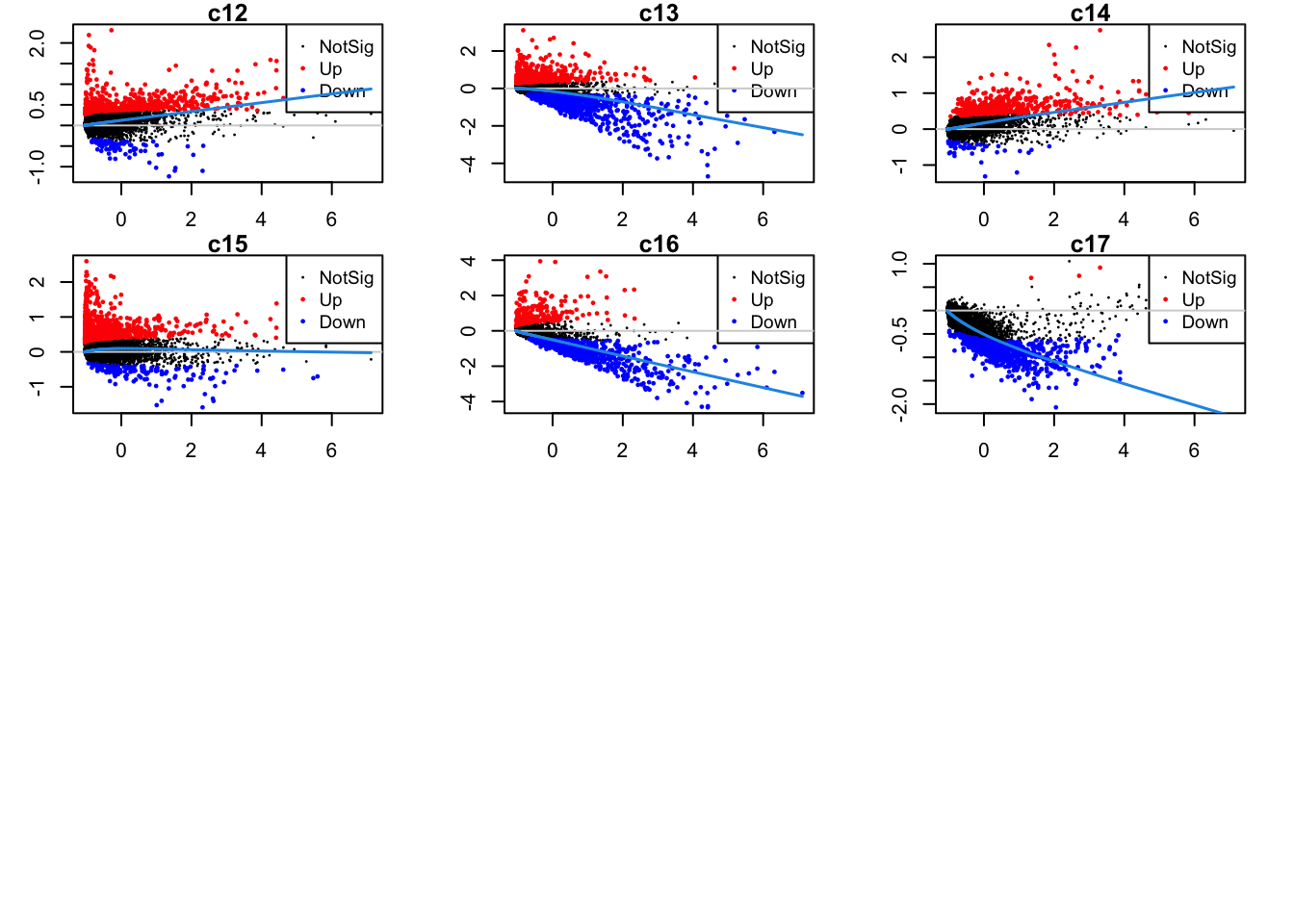
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Marker-gene dot plot
#Top 10 marker genes from limma analysis
DefaultAssay(paed_sub) <- "RNA"
contnames <- colnames(mycont)
top_markers <- NULL
n_markers <- 10
limma_markers_by_cluster <- vector("list", length = length(contnames))
names(limma_markers_by_cluster) <- contnames
for (i in seq_along(contnames)) {
top <- topTreat(tr, coef = i, n = Inf)
top <- top[top$logFC > 0.25, ] # Filter for significant markers
top_markers <- c(top_markers,
setNames(rownames(top)[1:n_markers],
rep(contnames[i], n_markers)))
markers <- rownames(top)
limma_markers_by_cluster[[contnames[i]]] <- markers
}
# Remove NA and duplicate markers
top_markers <- top_markers[!is.na(top_markers)]
top_markers <- top_markers[!duplicated(top_markers)]
cols <- paletteer::paletteer_d("pals::glasbey")[factor(names(top_markers))]
DotPlot(paed_sub,
features = unname(top_markers),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cols)) +
ggtitle("Top 10 cluster marker genes (Limma)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.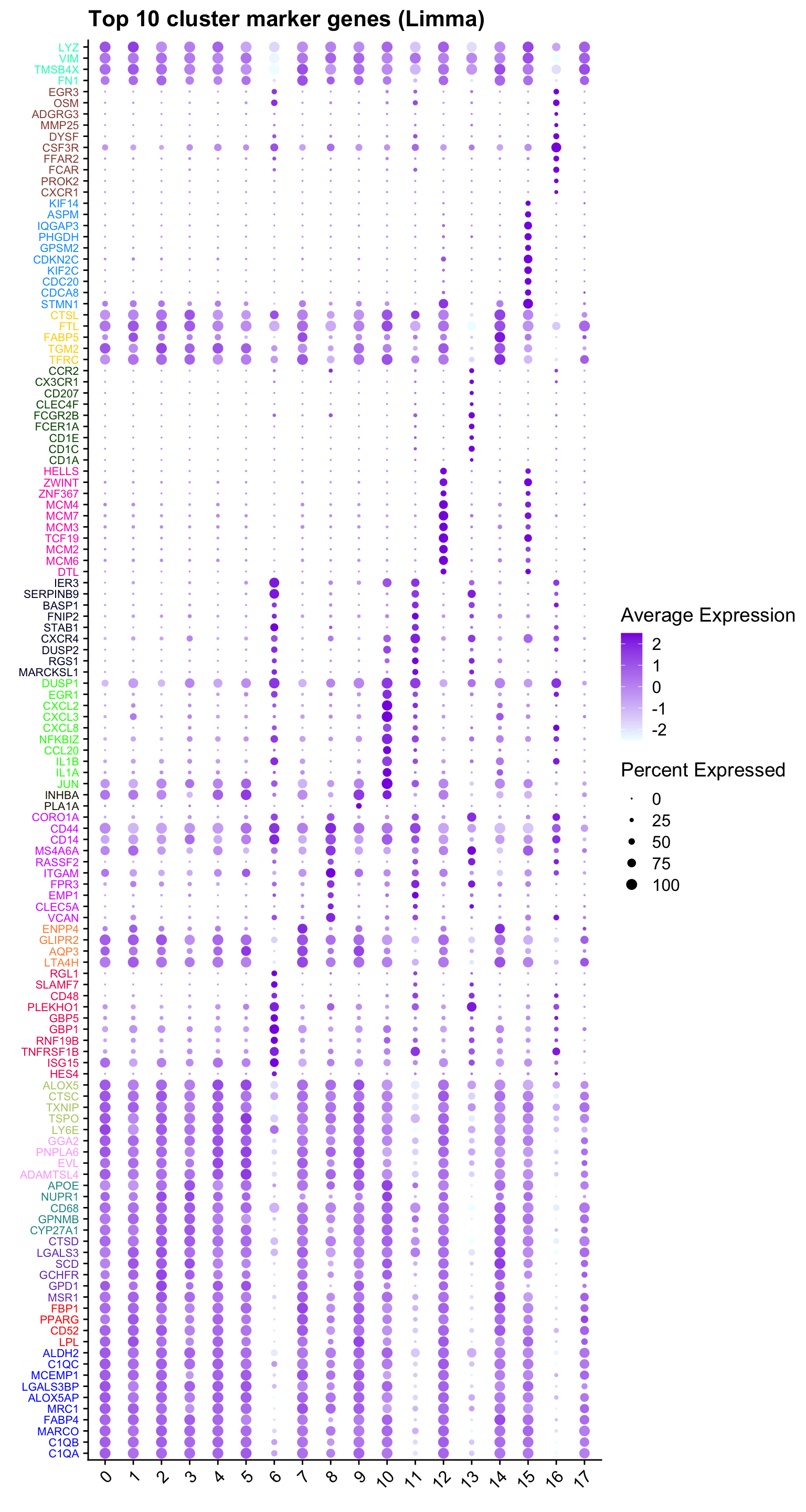
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.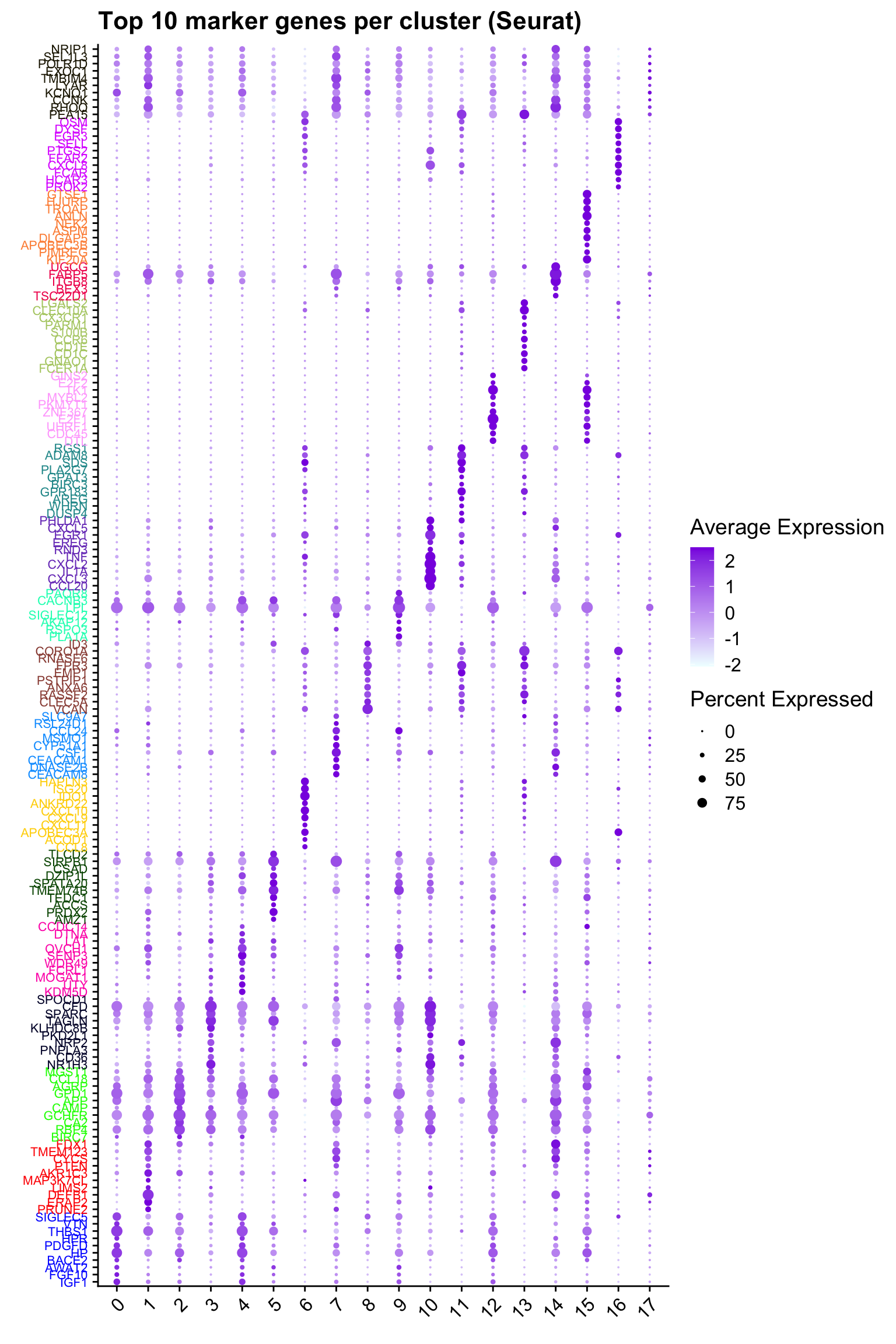
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
df_table <- as.data.frame(table(paed_sub$RNA_snn_res.0.1, paed_sub$predicted.ann_finest_level))
ggplot(df_table, aes(Var1, Freq, fill = Var2)) +
geom_bar(stat = "identity") +
labs(x = "RNA_snn_res.0.1", y = "Count", fill = "predicted ann_finest_level") +
theme_minimal() +
ggtitle("Stacked Bar Plot of predicted.ann_finest_level")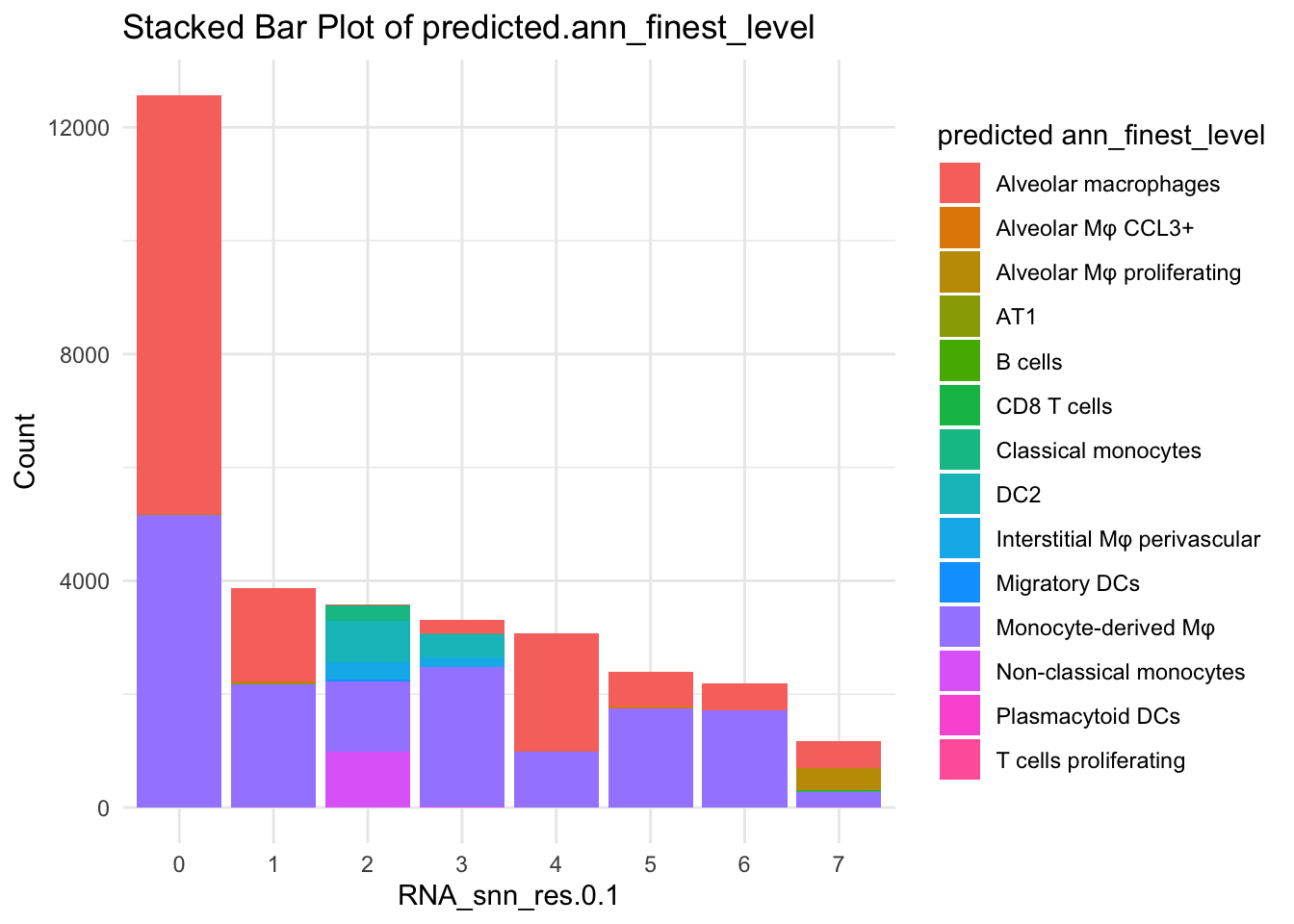
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
# Filter cells with >= 10 counts
valid_levels <- names(table(paed_sub$predicted.ann_finest_level)[table(paed_sub$predicted.ann_finest_level) >= 10])
paed_sub_filtered <- subset(paed_sub, subset = predicted.ann_finest_level %in% valid_levels)
# Stacked bar plot with filtered cells
df_table_filtered <- as.data.frame(table(paed_sub_filtered$RNA_snn_res.0.1, paed_sub_filtered$predicted.ann_finest_level))
ggplot(df_table_filtered, aes(x = Var1, y = Freq, fill = Var2)) +
geom_bar(stat = "identity") +
labs(x = "RNA_snn_res.0.1", y = "Count", fill = "predicted ann_finest_level") +
theme_minimal() +
ggtitle("Stacked Bar Plot of predicted.ann_finest_level")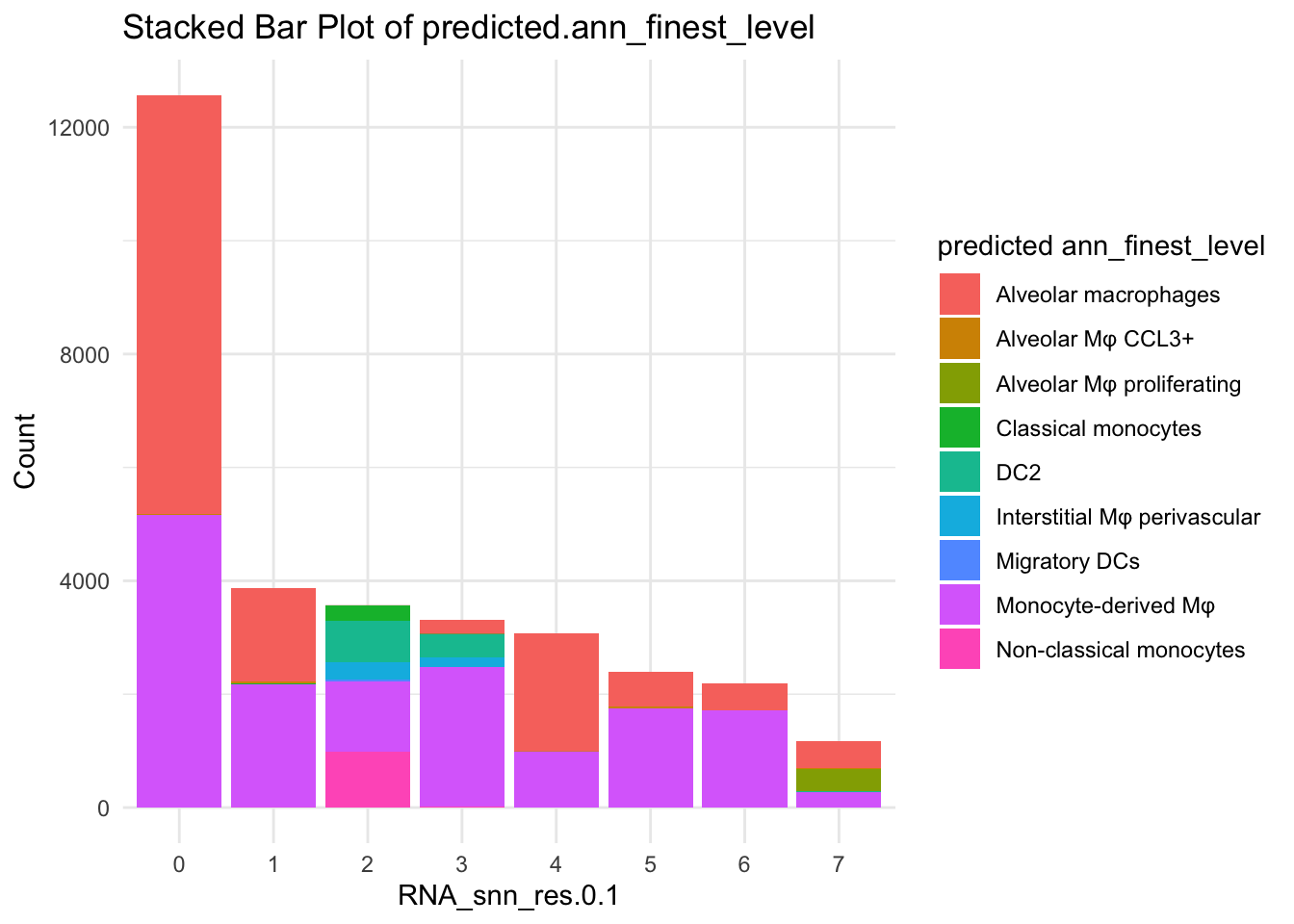
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Overlapping and Unique markers between Seurat and Limma approach
limma_markers_by_cluster <- setNames(limma_markers_by_cluster, gsub("^c", "", names(limma_markers_by_cluster)))
limma_markers_df <- do.call(rbind, lapply(names(limma_markers_by_cluster), function(cluster) {
data.frame(cluster = cluster, gene = limma_markers_by_cluster[[cluster]], stringsAsFactors = FALSE)
}))
seurat_markers_df <- paed_sub.markers[, c("cluster", "gene")]
marker_comparison_list <- list()
for (cluster in unique(names(limma_markers_by_cluster))) {
limma_genes <- limma_markers_df$gene[limma_markers_df$cluster == cluster]
seurat_genes <- seurat_markers_df$gene[seurat_markers_df$cluster == cluster]
overlap <- intersect(limma_genes, seurat_genes)
unique_limma <- setdiff(limma_genes, seurat_genes)
unique_seurat <- setdiff(seurat_genes, limma_genes)
marker_comparison_list[[cluster]] <- data.frame(
Cluster = cluster,
Category = c("Overlapping", "Unique Limma", "Unique Seurat"),
Count = c(length(overlap), length(unique_limma), length(unique_seurat))
)
}
marker_comparison_df <- do.call(rbind, marker_comparison_list)
marker_comparison_df$Cluster <- factor(marker_comparison_df$Cluster, levels = sort(as.numeric(unique(marker_comparison_df$Cluster))))
ggplot(marker_comparison_df, aes(x = Cluster, y = Count, fill = Category)) +
geom_bar(stat = "identity", position = "stack") +
theme_minimal() +
labs(title = "Comparison of Marker Genes by Cluster (lfc: 0.25)",
y = "Number of Genes",
x = "Cluster") +
scale_fill_manual(values = c("Overlapping" = "darkgreen", "Unique Limma" = "cornflowerblue", "Unique Seurat" = "darkorchid1"))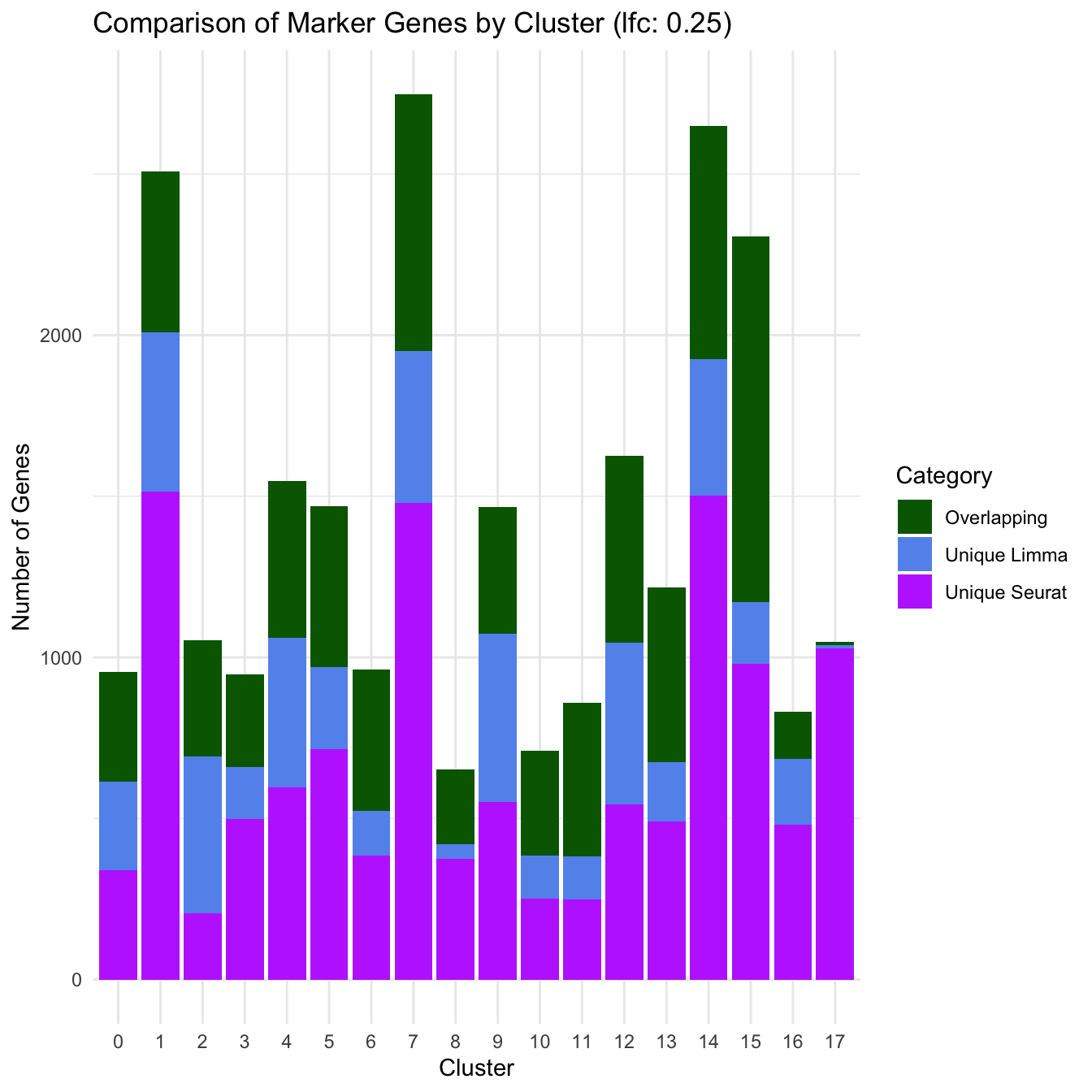
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Update Macro labels
cell_labels <- readxl::read_excel(here("data/Cell_labels_Mel_v2/earlyAIR_BAL_macrophage_reclustering_annotations_16.07.24.xlsx"))
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
paed_sub <- RenameIdents(paed_sub, new_cluster_names)
paed_sub@meta.data$cell_labels_v2 <- Idents(paed_sub)
DimPlot(paed_sub, reduction = "umap.new", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated subclustering labels"))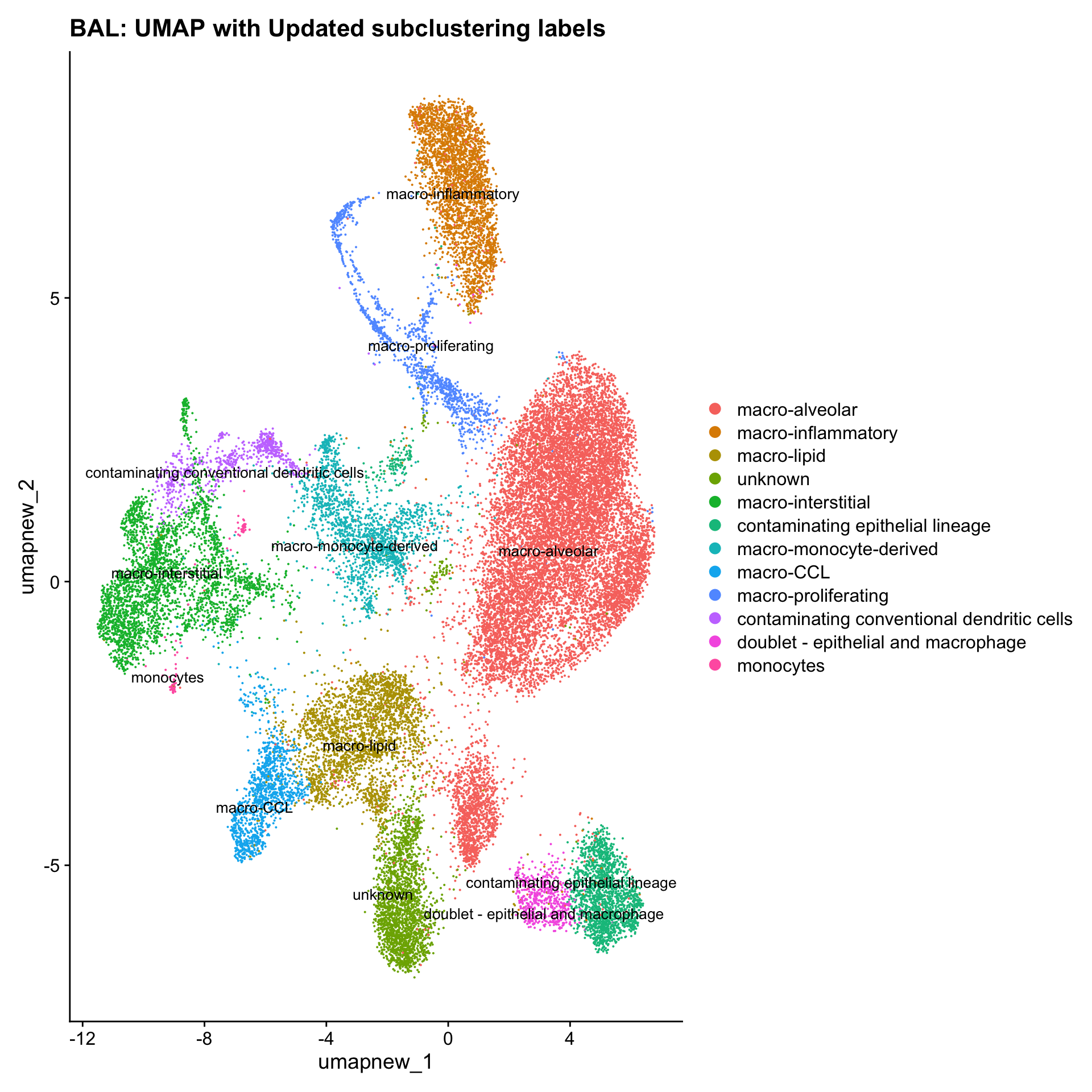
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Save subclustered SEU object (Macro Population)
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".macro_population.subclusters.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Excluding contaminating labels
idx <- which(grepl("^contaminating", Idents(paed_sub)))
paed_clean <- paed_sub[, -idx]
DimPlot(paed_clean, reduction = "umap.new", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ":Updated subclustering (clean)"))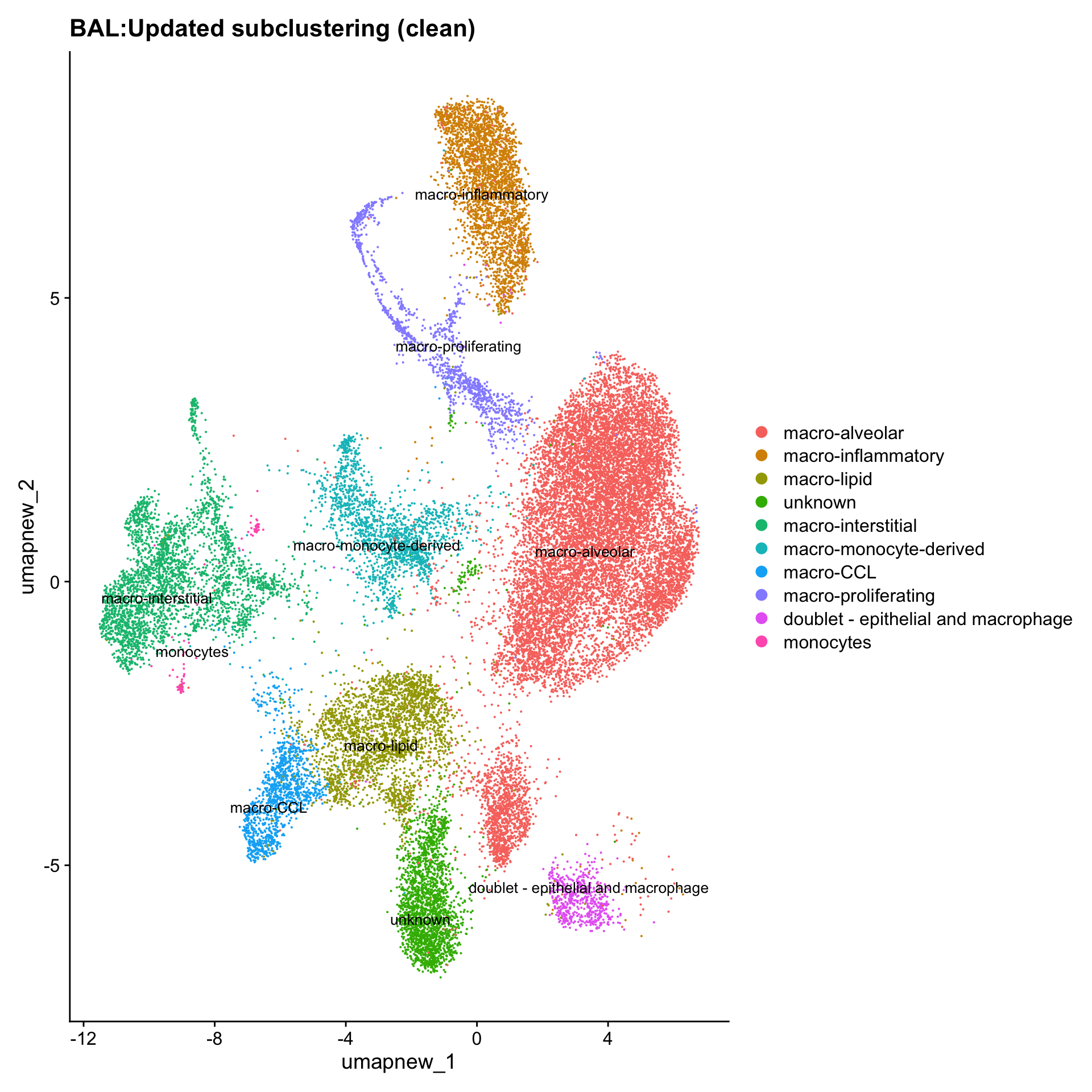
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
paed_clean <- paed_clean %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()Normalizing layer: countsFinding variable features for layer countsCentering and scaling data matrixWarning: Different features in new layer data than already exists for
scale.dataPC_ 1
Positive: CD52, FABP4, MRC1, FBP1, MCEMP1, LTA4H, CRIP1, LPL, MSR1, ABCG1
PECAM1, CYP27A1, GPNMB, PPARG, CD9, GPD1, PCOLCE2, IGFBP2, ANXA1, SCD
LYZ, MME, ANXA2, AMIGO2, LGALS3, C8B, EVL, TSPAN3, PDLIM1, GCHFR
Negative: SOCS3, IER3, SERPINB9, SOD2, NINJ1, ZFP36, SPHK1, MARCKS, IL4I1, GPR132
LILRB2, PFKFB3, NR4A1, C15orf48, IDO1, VEGFA, CCL3, NFKBIA, JUNB, ICAM1
STAB1, DUSP1, PIM3, SAT1, NR4A3, TNFRSF1B, TIMP1, HAPLN3, ATF3, CD300E
PC_ 2
Positive: TYMS, MKI67, KIFC1, TOP2A, CENPM, HIST1H1B, RRM2, BIRC5, PCLAF, TPX2
CDK1, SPC24, FOXM1, MYBL2, TK1, ANLN, CDCA5, CDT1, UHRF1, ASF1B
PRC1, CEP55, AURKB, NCAPG, NDC80, TCF19, GTSE1, CIT, DTL, HJURP
Negative: GPD1, C5AR1, MSR1, CD9, TGM2, TFRC, GSN, TYMP, AQP3, ABCG1
PCOLCE2, MRC1, EVL, CYP27A1, MCEMP1, FTL, HMOX1, SLC19A3, INHBA, ADGRE3
GLRX, RGCC, FCGR3A, ACE, CFD, HBEGF, SQSTM1, CACNB3, TMEM273, LTA4H
PC_ 3
Positive: THBS1, FGL2, TMEM273, CISH, PLAC8, MCEMP1, ISG15, MRC1, HP, PLEKHO1
NFE2, PDGFD, IGF1, ADGRE3, CD101, TNFSF10, CD1D, IFI6, IL3RA, AWAT2
CD69, ITGAM, ECSCR, FGF10, MS4A6A, IFITM3, MYB, MEFV, PTGER3, AQP3
Negative: CSTB, CTSL, TFRC, SCD, LIPA, CXCL3, NR1H3, ABCA1, GPNMB, CD83
CD36, APOC1, CXCL2, FTL, AQP9, PLPP3, RMDN3, NRP2, CCL20, LGALS3
GCHFR, BCL2A1, ACSL1, LGALS1, CTSB, CCL18, IL1A, PHLDA1, MACC1, SDCBP
PC_ 4
Positive: MS4A6A, GLIPR1, TPM4, CYBB, FPR3, CMTM6, EIF1, CLIC4, CD47, AHR
TMEM176B, TMEM123, TMEM176A, SNX2, EVI2B, IFNGR1, RNF13, DEFB1, DNAJA1, MPEG1
STT3B, FAM49B, LAMP2, ANXA2, HACD4, EIF4G2, TRAM1, SPPL2A, SNX3, SGMS2
Negative: CFD, JUN, SQSTM1, NUPR1, TAGLN, CYP27A1, APOE, TNFAIP2, CCL20, PLAUR
TNF, IL1A, TGM2, BCAR1, G0S2, CXCL8, PPP1R15A, CD82, CD83, GPD1
GSN, CCL3, CCL4, SDC4, PKD2L1, INHBA, FGR, CD276, BTG2, CXCL2
PC_ 5
Positive: FPR3, EMP1, RASSF2, LMNA, CLEC10A, EMP3, CLEC5A, A2M, RNASE1, CXCR4
LGALS1, CST7, ANXA6, BCAT1, F13A1, MYADM, SNCA, ADAM8, PAPSS2, PMP22
NRP2, RNASE6, GPAT3, CORO1A, S100A10, ARL4C, ARHGEF40, GPR183, CCR7, CRIP1
Negative: CXCL10, APOBEC3A, S100A9, ISG15, CCL8, GBP1, CXCL11, IFIT2, ISG20, IDO1
GLUL, GBP5, MX1, IFI6, ACOD1, LILRA5, IFIT3, RSAD2, SERPING1, WARS
CD38, GCH1, C8B, IL1RN, GBP2, CALHM6, IL1B, PCOLCE2, LILRB1, TNFSF10 paed_clean <- RunUMAP(paed_clean, dims = 1:30, reduction = "pca", reduction.name = "umap.clean")11:35:11 UMAP embedding parameters a = 0.9922 b = 1.112Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'11:35:11 Read 29638 rows and found 30 numeric columns11:35:11 Using Annoy for neighbor search, n_neighbors = 30Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'11:35:11 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
11:35:13 Writing NN index file to temp file /var/folders/q8/kw1r78g12qn793xm7g0zvk94x2bh70/T//Rtmpx9umSA/filedfb4ba8c4e7
11:35:13 Searching Annoy index using 1 thread, search_k = 3000
11:35:18 Annoy recall = 100%
11:35:18 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
11:35:19 Initializing from normalized Laplacian + noise (using RSpectra)
11:35:20 Commencing optimization for 200 epochs, with 1283702 positive edges
11:35:27 Optimization finishedDimPlot(paed_clean, reduction = "umap.clean", group.by = "cell_labels_v2",raster = FALSE, repel = TRUE, label = TRUE, label.size = 4.5) + ggtitle(paste0(tissue, ": Updated subclustering (clean)"))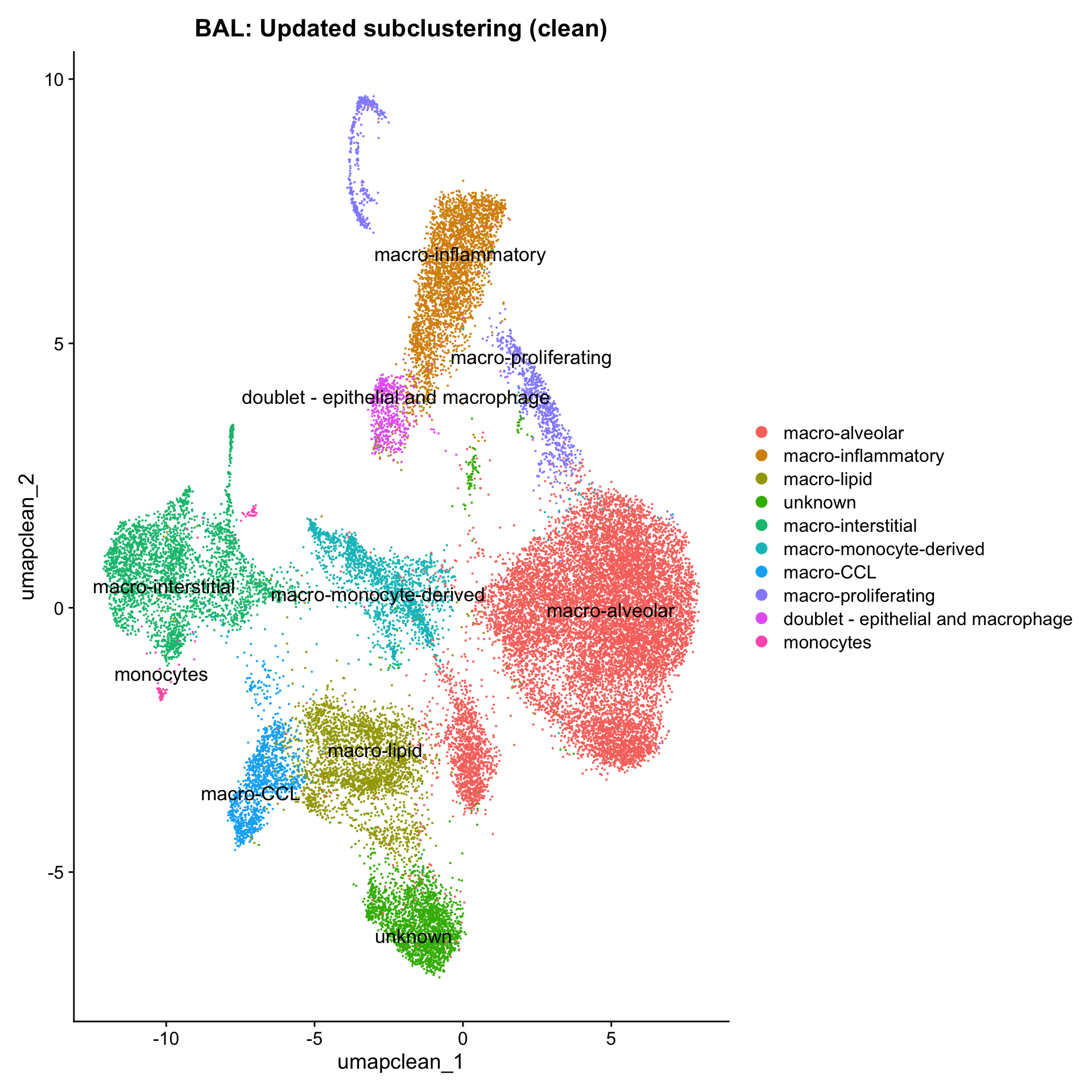
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Reclustering Tcell polulation
This includes CD4 T cell, CD8 T cell, NK cell, NK-T cell, proliferating or cycling T/NK cell.
The marker genes for this reclustering can be found
idx <- which(Idents(seu_obj) %in% c("CD4 T cells", "CD8 T cells", "NK-T cells", "proliferating T/NK", "cycling T cells"))
paed_sub <- seu_obj[,idx]
mito_genes <- grep("^MT-", rownames(paed_sub), value = TRUE)
paed_sub <- subset(paed_sub, features = setdiff(rownames(paed_sub), mito_genes))
paed_subAn object of class Seurat
17518 features across 4877 samples within 1 assay
Active assay: RNA (17518 features, 1990 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, umap, umap.unintegratedpaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9386
Number of communities: 5
Elapsed time: 2 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9134
Number of communities: 8
Elapsed time: 2 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8944
Number of communities: 10
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8777
Number of communities: 12
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8625
Number of communities: 13
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8499
Number of communities: 13
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8376
Number of communities: 14
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8279
Number of communities: 16
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8186
Number of communities: 16
Elapsed time: 1 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4877
Number of edges: 185348
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8096
Number of communities: 17
Elapsed time: 1 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
clustree(paed_sub, prefix = "RNA_snn_res.")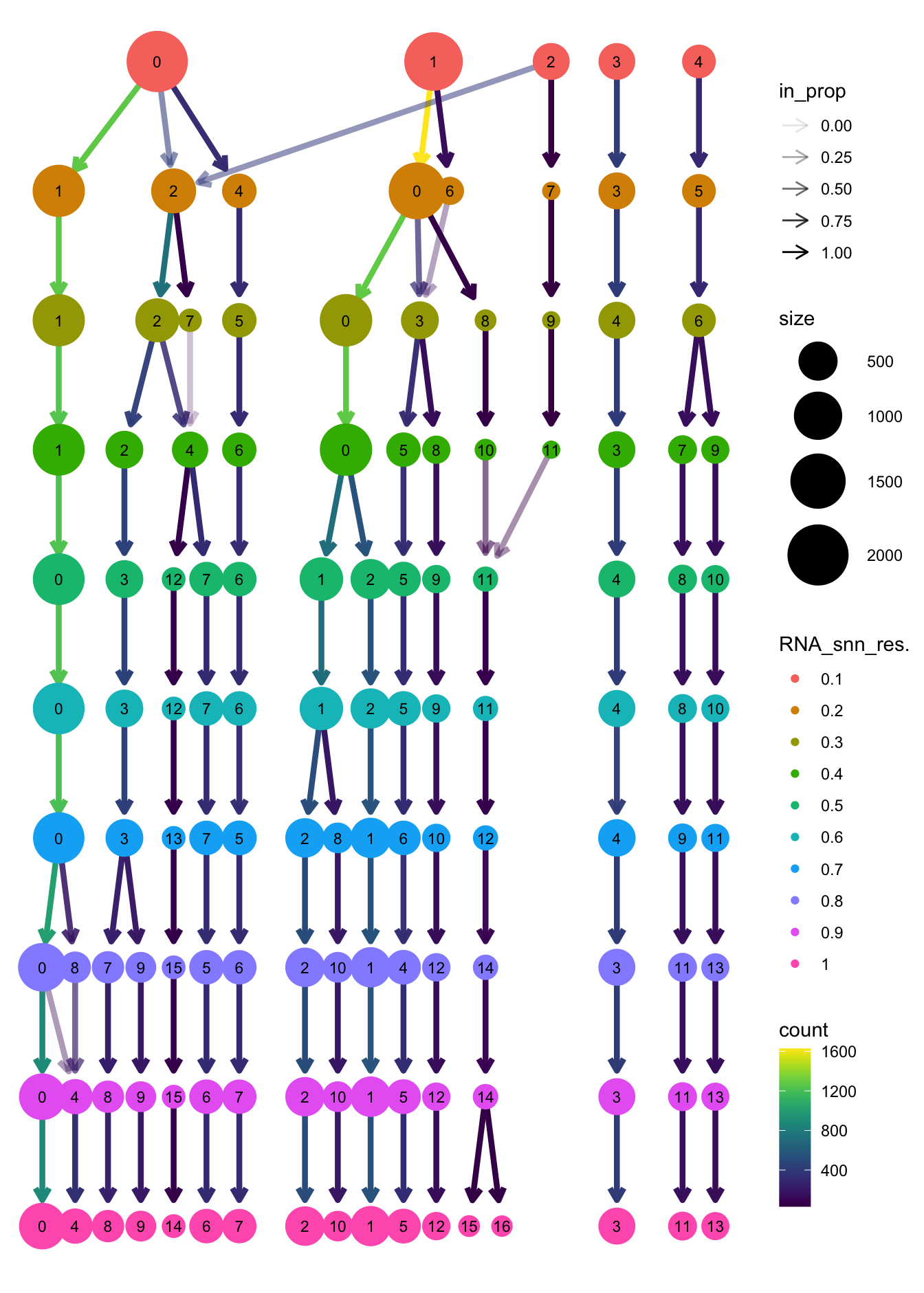
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(paed_sub$RNA_snn_res.0.4)
paed_sub$RNA_snn_res.0.4 <- factor(paed_sub$RNA_snn_res.0.4, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
paed_sub$cluster <- paed_sub$RNA_snn_res.0.4
Idents(paed_sub) <- paed_sub$clusterpaed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3Calculating cluster 4Calculating cluster 5Calculating cluster 6Calculating cluster 7Calculating cluster 8Calculating cluster 9Calculating cluster 10Calculating cluster 11paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster [1] "CD4" "CCL5" "TCF7" "NMUR1" "AREG" "MAF" "GZMB" "CD79A"
[9] "CXCR5" "TYMS" "SCART1" "CX3CR1"Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, ncol = 2, label = TRUE)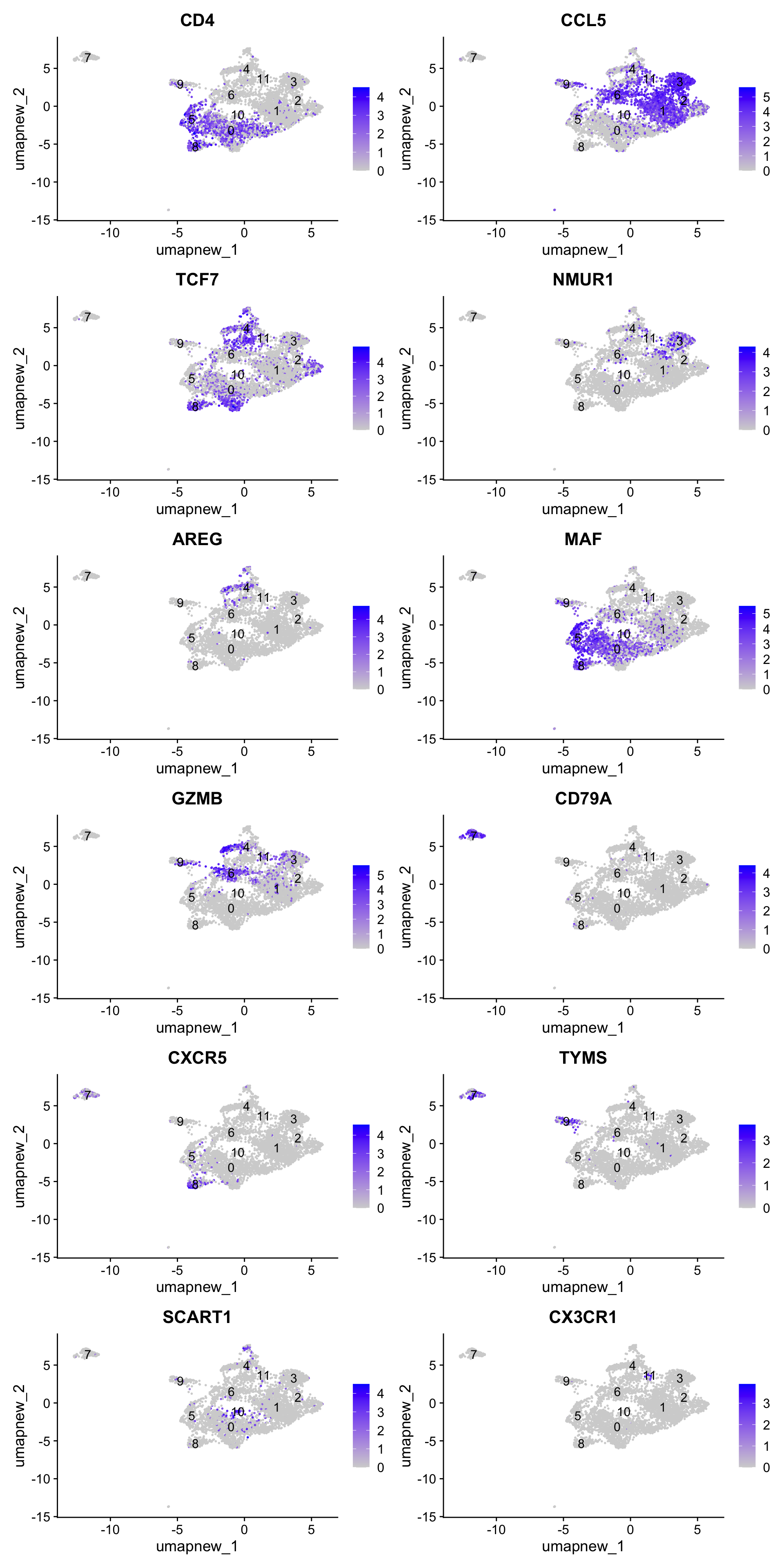
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.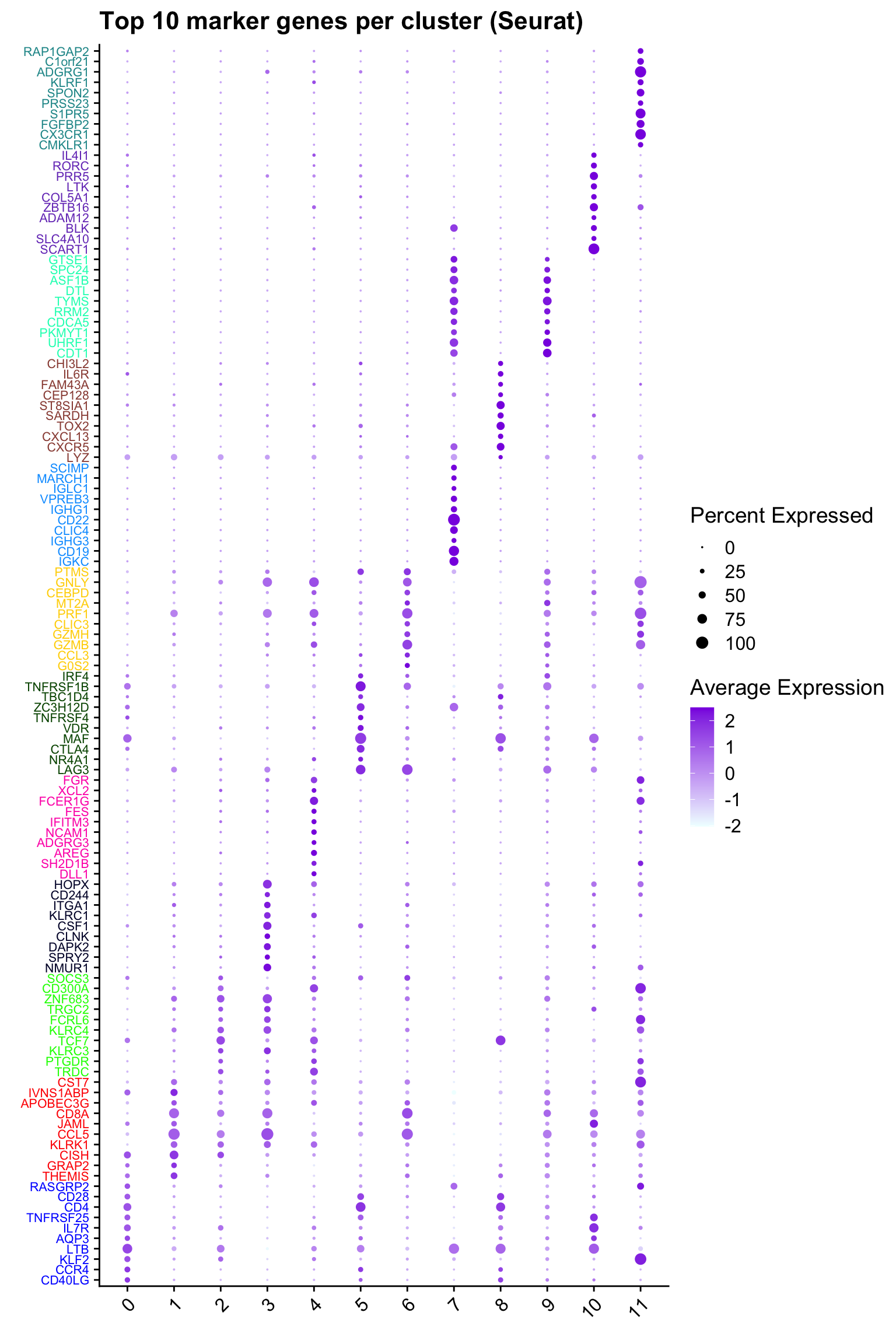
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_Tcell_population.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
if (!file.exists(file_name)) {
write.csv(cluster_data, file = file_name)
}
}Update T cell subclustering labels
cell_labels <- readxl::read_excel(here("data/Cell_labels_Mel_v2/earlyAIR_NB_BB_BAL_T-NK_annotations_16.07.24.xlsx"), sheet = "BAL")
new_cluster_names <- cell_labels %>%
dplyr::select(cluster, annotation) %>%
deframe()
paed_sub <- RenameIdents(paed_sub, new_cluster_names)
paed_sub@meta.data$cell_labels_v2 <- Idents(paed_sub)
DimPlot(paed_sub, reduction = "umap.new", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": UMAP with Updated cell types"))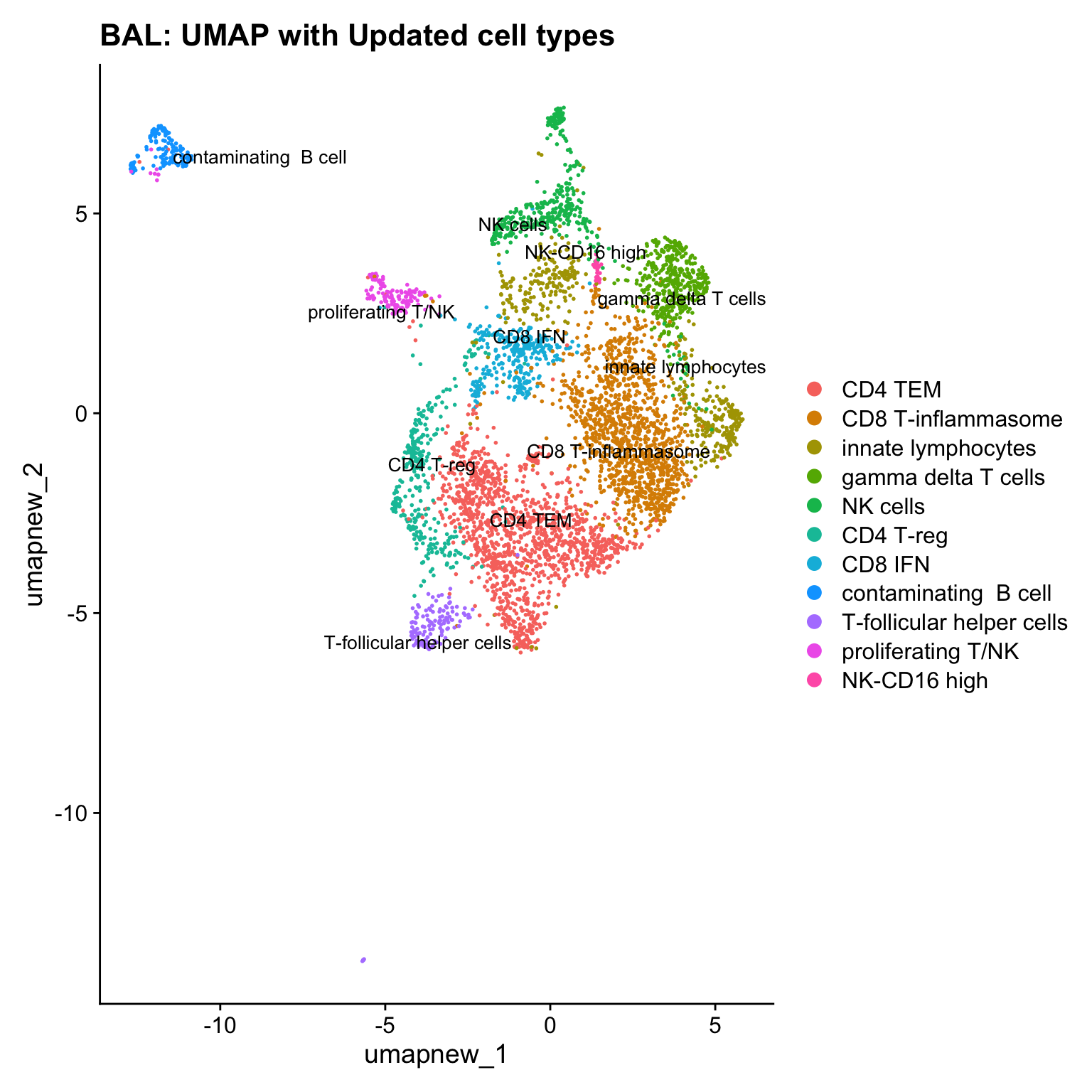
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Excluding contaminating labels
idx <- which(grepl("^contaminating", Idents(paed_sub)))
paed_clean <- paed_sub[, -idx]
DimPlot(paed_clean, reduction = "umap.new", raster = FALSE, repel = TRUE, label = TRUE, label.size = 3.5) + ggtitle(paste0(tissue, ": Updated subclustering (clean)"))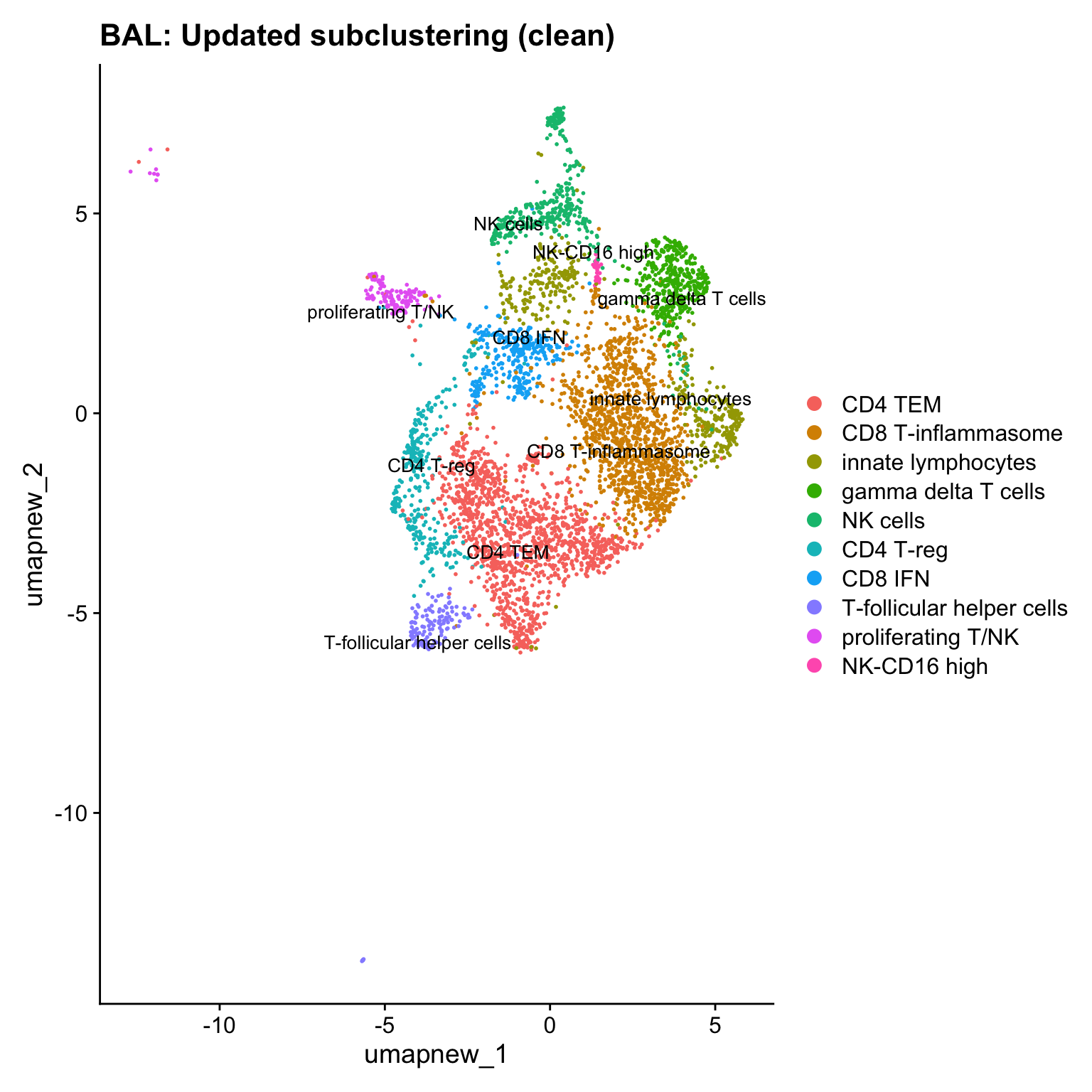
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
paed_clean <- paed_clean %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()Normalizing layer: countsFinding variable features for layer countsCentering and scaling data matrixWarning: Different features in new layer data than already exists for
scale.dataPC_ 1
Positive: IL7R, TCF7, CISH, PTGDR, KLRK1, TSC22D3, KLRC3, AOAH, TRGC2, FCRL6
OBSCN, TRGC1, PLXDC1, KLRC4, CCDC141, SCML4, NMUR1, SPRY2, PTGER2, TRDC
TC2N, NELL2, FCGBP, ADAMTS10, ABCB1, LEF1, TRGJP2, TTN, GCSAM, PIK3R1
Negative: TYMS, CDT1, UHRF1, RRM2, MKI67, HIST1H1B, KIFC1, ASF1B, MYBL2, AURKB
ZWINT, TOP2A, TK1, PKMYT1, CDCA5, SPC24, DTL, E2F1, FOXM1, BIRC5
PCLAF, STMN1, HIST1H2BH, ESPL1, TPX2, NUSAP1, GTSE1, ASPM, CDK1, E2F2
PC_ 2
Positive: MAF, CD4, SPOCK2, LTB, CD28, CCR4, ZC3H12D, CD6, CTLA4, TNFRSF25
CD5, TNFRSF4, PIM2, AQP3, PBXIP1, CD40LG, ICOS, COL5A3, TMEM173, CTSH
IL7R, CCR6, IL6R, TNFRSF1B, ADAM19, NPDC1, IL2RA, CERK, SLAMF1, TBC1D4
Negative: NKG7, CTSW, GNLY, CCL5, PRF1, ZNF683, KLRD1, CD8A, MATK, ITGAX
HOPX, KLRC4, CD7, KLRK1, GZMB, NCR1, KLRC3, PIK3AP1, KLRC1, NMUR1
RIN3, FCRL6, AOAH, FGR, TRDC, KIR2DL4, GZMA, IL2RB, ITM2C, SPRY2
PC_ 3
Positive: FURIN, DUSP2, SRGN, GZMB, TNFRSF18, NR4A1, IL2RB, FOSL2, GNLY, CD38
DDIT4, CD7, VDR, ZFP36, ISG15, BCL3, IFI6, SOCS3, FOS, AREG
JUNB, IFITM3, HAPLN3, IRF8, SBNO2, SH2D2A, NR4A2, ISG20, NFKBIA, NR4A3
Negative: CISH, VIM, HIST1H1C, ANXA1, RRM2, CRIP1, BBC3, HIST1H1B, GRAP2, PLP2
TOP2A, ASPM, TRIB3, MKI67, TYMS, CCL5, HIST1H1D, KIFC1, NCAPG, ANKRD28
HIST1H2BH, KDM5D, STMN1, HJURP, FAM111A, CKS1B, TC2N, CDK1, UTY, JAML
PC_ 4
Positive: LAG3, CCL5, CXCR6, CD8A, GZMA, CCR5, IL32, PTMS, TIGIT, CCL4
CSF1, TRBC2, GPR25, ITGA1, DUSP4, ABI3, DAPK2, PRDM1, CCL4L2, TNFRSF1B
LBH, PLAAT4, PHLDA1, PDCD1, FASLG, CD6, S100A4, JUN, CCL3, GZMH
Negative: TCF7, AREG, PLAC8, DLL1, KLF2, CD300A, IL7R, TRDC, NCAM1, IFITM3
LTB, SH2D1B, TXK, KIT, FES, TNFRSF18, RIPOR2, ITGAM, PTGDR, SELL
LIF, ADGRG3, SLC16A3, XCL2, FGR, IRF8, S1PR1, KLRF1, SORL1, BHLHE40
PC_ 5
Positive: CXCR5, TOX2, TCF7, IGHM, FCMR, POU2AF1, CHI3L2, GNG4, ID3, CD7
KIAA1324, SARDH, ITGAX, SIRPG, NMUR1, RTP5, CDK5R1, ZNF703, IL21, ST8SIA1
TBC1D4, FCRL6, CXXC5, TSPOAP1, MAGEH1, FAM43A, CXCL13, KCNK5, LTBP3, SPRY2
Negative: CISH, VIM, ISG15, ANXA1, CRIP1, RSAD2, OAS3, MX1, CMPK2, ADAM19
LGALS1, IFI44L, OAS1, IFI6, IFIT1, MYADM, XAF1, LY6E, TYMP, CCR5
PRF1, BHLHE40, GBP1, OAS2, OASL, IFI44, USP18, GZMB, IRF7, APOBEC3G paed_clean <- RunUMAP(paed_clean, dims = 1:30, reduction = "pca", reduction.name = "umap.clean")11:36:11 UMAP embedding parameters a = 0.9922 b = 1.112Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'11:36:11 Read 4724 rows and found 30 numeric columns11:36:11 Using Annoy for neighbor search, n_neighbors = 30Found more than one class "dist" in cache; using the first, from namespace 'spam'Also defined by 'BiocGenerics'11:36:11 Building Annoy index with metric = cosine, n_trees = 500% 10 20 30 40 50 60 70 80 90 100%[----|----|----|----|----|----|----|----|----|----|**************************************************|
11:36:11 Writing NN index file to temp file /var/folders/q8/kw1r78g12qn793xm7g0zvk94x2bh70/T//Rtmpx9umSA/filedfb2aedb2f9
11:36:11 Searching Annoy index using 1 thread, search_k = 3000
11:36:12 Annoy recall = 100%
11:36:12 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
11:36:13 Initializing from normalized Laplacian + noise (using RSpectra)
11:36:13 Commencing optimization for 500 epochs, with 194062 positive edges
11:36:16 Optimization finishedDimPlot(paed_clean, reduction = "umap.clean", group.by = "cell_labels_v2",raster = FALSE, repel = TRUE, label = TRUE, label.size = 4.5) + ggtitle(paste0(tissue, ": Updated subclustering (clean)"))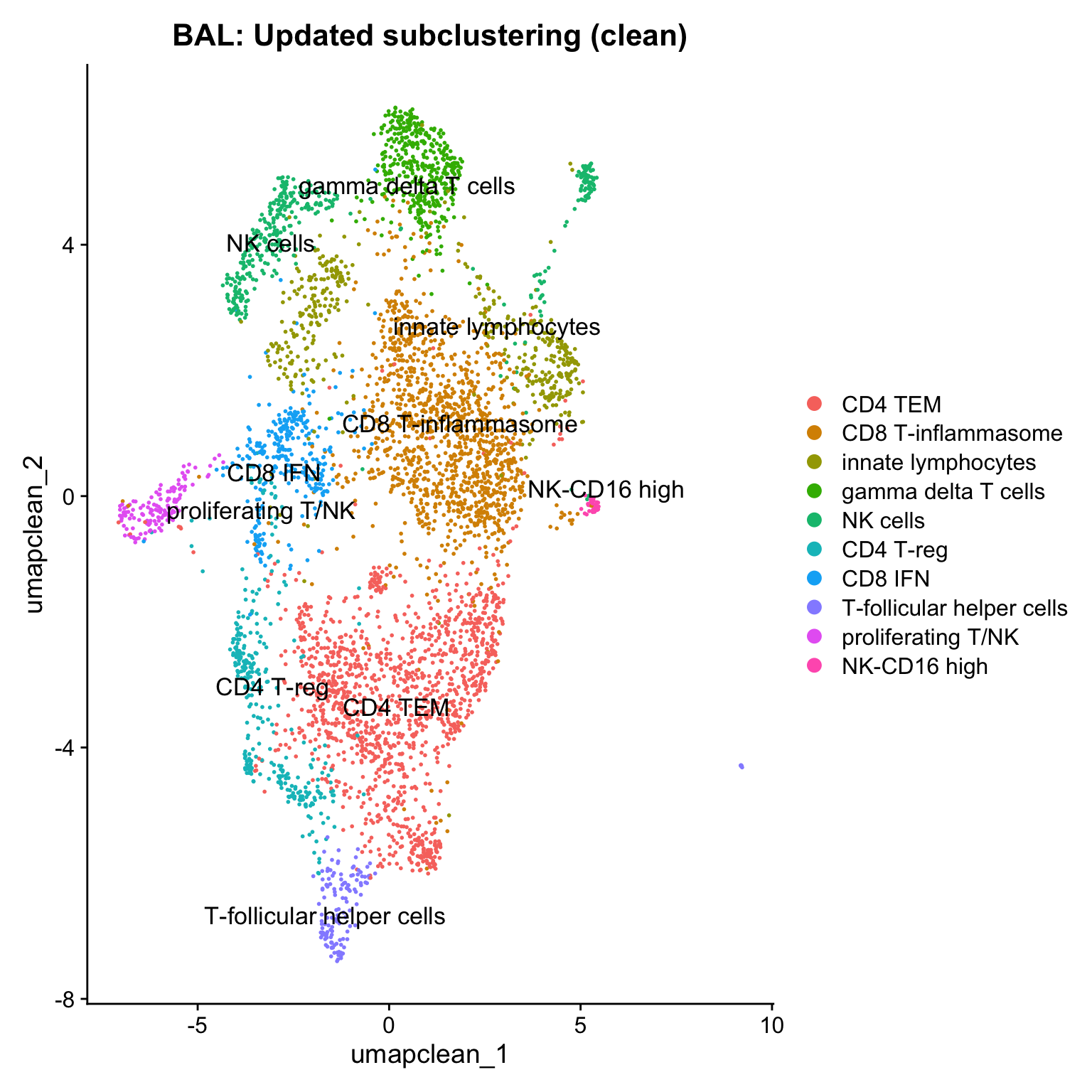
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Save subclustered SEU object
out3 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Tcell_population.subclusters.SEU.rds"))
#dir.create(out2)
if (!file.exists(out3)) {
saveRDS(paed_sub, file = out3)
}Reclustering Bcell polulation
Here is the link to marker gene analysis of Macrophages in BAL (without ambient removal) BAL_Bcell_res.0.4
idx <- which(Idents(seu_obj) %in% "B cells")
paed_sub <- seu_obj[,idx]
paed_subAn object of class Seurat
17529 features across 1755 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, umap, umap.unintegratedpaed_sub <- paed_sub %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
paed_sub <- RunUMAP(paed_sub, dims = 1:30, reduction = "pca", reduction.name = "umap.new")
meta_data_columns <- colnames(paed_sub@meta.data)
columns_to_remove <- grep("^RNA_snn_res", meta_data_columns, value = TRUE)
paed_sub@meta.data <- paed_sub@meta.data[, !(colnames(paed_sub@meta.data) %in% columns_to_remove)]
resolutions <- seq(0.1, 1, by = 0.1)
paed_sub <- FindNeighbors(paed_sub, reduction = "pca", dims = 1:30)
paed_sub <- FindClusters(paed_sub, resolution = resolutions, algorithm = 3)Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.9165
Number of communities: 2
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8718
Number of communities: 3
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8350
Number of communities: 3
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.8076
Number of communities: 4
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7838
Number of communities: 5
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7614
Number of communities: 5
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7393
Number of communities: 5
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7206
Number of communities: 7
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.7037
Number of communities: 7
Elapsed time: 0 seconds
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 1755
Number of edges: 80164
Running smart local moving algorithm...
Maximum modularity in 10 random starts: 0.6870
Number of communities: 8
Elapsed time: 0 secondsDimHeatmap(paed_sub, dims = 1:10, cells = 500, balanced = TRUE)
clustree(paed_sub, prefix = "RNA_snn_res.")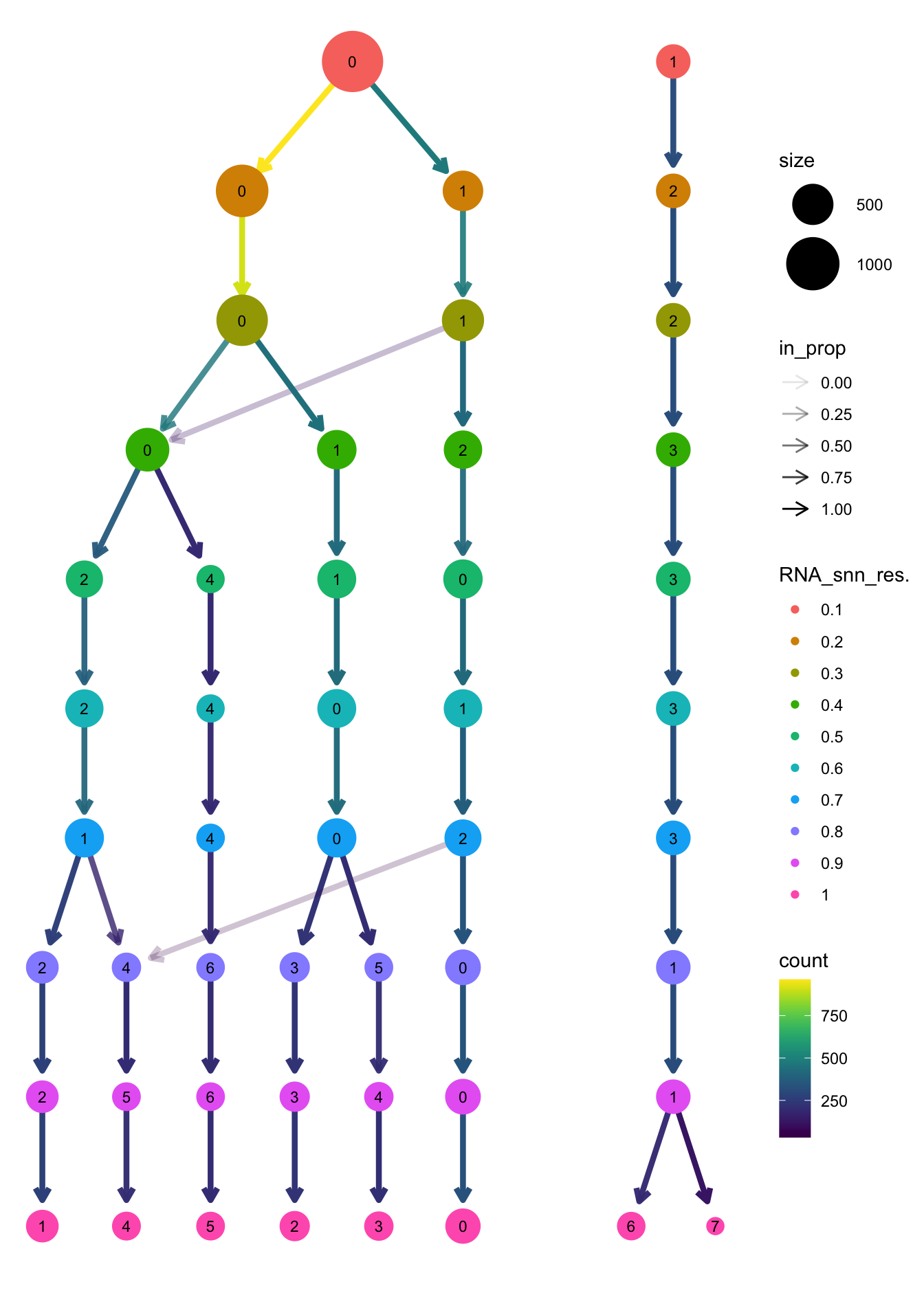
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
opt_res <- "RNA_snn_res.0.4"
n <- nlevels(paed_sub$RNA_snn_res.0.4)
paed_sub$RNA_snn_res.0.4 <- factor(paed_sub$RNA_snn_res.0.4, levels = seq(0,n-1))
paed_sub$seurat_clusters <- NULL
paed_sub$cluster <- paed_sub$RNA_snn_res.0.4
Idents(paed_sub) <- paed_sub$clusterpaed_sub.markers <- FindAllMarkers(paed_sub, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)Calculating cluster 0Calculating cluster 1Calculating cluster 2Calculating cluster 3paed_sub.markers %>%
group_by(cluster) %>% unique() %>%
top_n(n = 5, wt = avg_log2FC) -> top5
paed_sub.markers %>%
group_by(cluster) %>%
slice_head(n=1) %>%
pull(gene) -> best.wilcox.gene.per.cluster
best.wilcox.gene.per.cluster[1] "ITGAX" "DUSP4" "IGHD" "MEF2B"Feature plot shows the expression of top marker genes per cluster.
FeaturePlot(paed_sub,features=best.wilcox.gene.per.cluster, reduction = 'umap.new', raster = FALSE, ncol = 2, label = TRUE)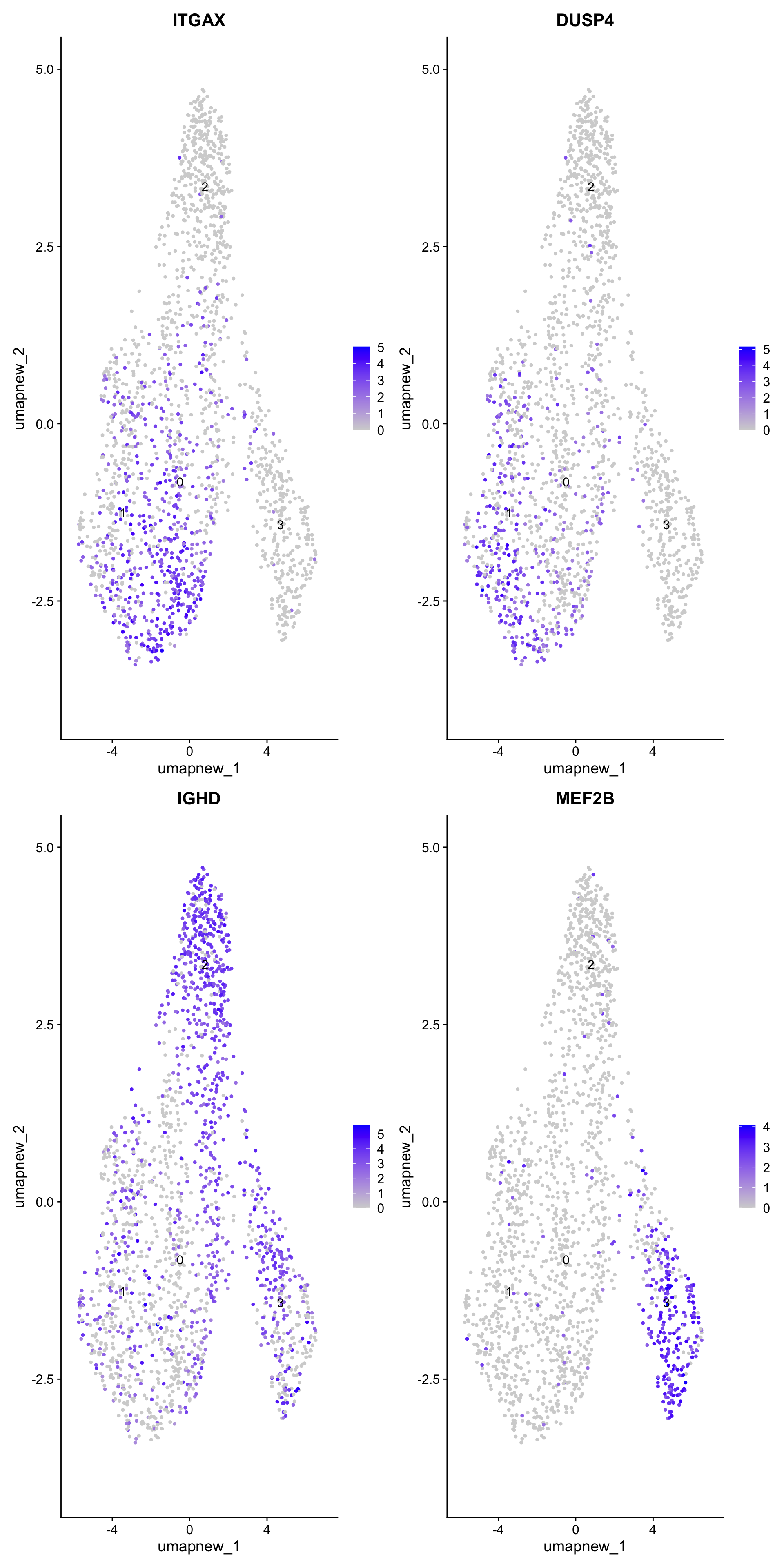
| Version | Author | Date |
|---|---|---|
| 07af966 | Gunjan Dixit | 2024-09-25 |
Top 10 marker genes from Seurat
## Seurat top markers
top10 <- paed_sub.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) %>%
ungroup() %>%
distinct(gene, .keep_all = TRUE) %>%
arrange(cluster, desc(avg_log2FC))
cluster_colors <- paletteer::paletteer_d("pals::glasbey")[factor(top10$cluster)]
DotPlot(paed_sub,
features = unique(top10$gene),
group.by = opt_res,
cols = c("azure1", "blueviolet"),
dot.scale = 3, assay = "RNA") +
RotatedAxis() +
FontSize(y.text = 8, x.text = 12) +
labs(y = element_blank(), x = element_blank()) +
coord_flip() +
theme(axis.text.y = element_text(color = cluster_colors)) +
ggtitle("Top 10 marker genes per cluster (Seurat)")Warning: Scaling data with a low number of groups may produce misleading
resultsWarning: Vectorized input to `element_text()` is not officially supported.
ℹ Results may be unexpected or may change in future versions of ggplot2.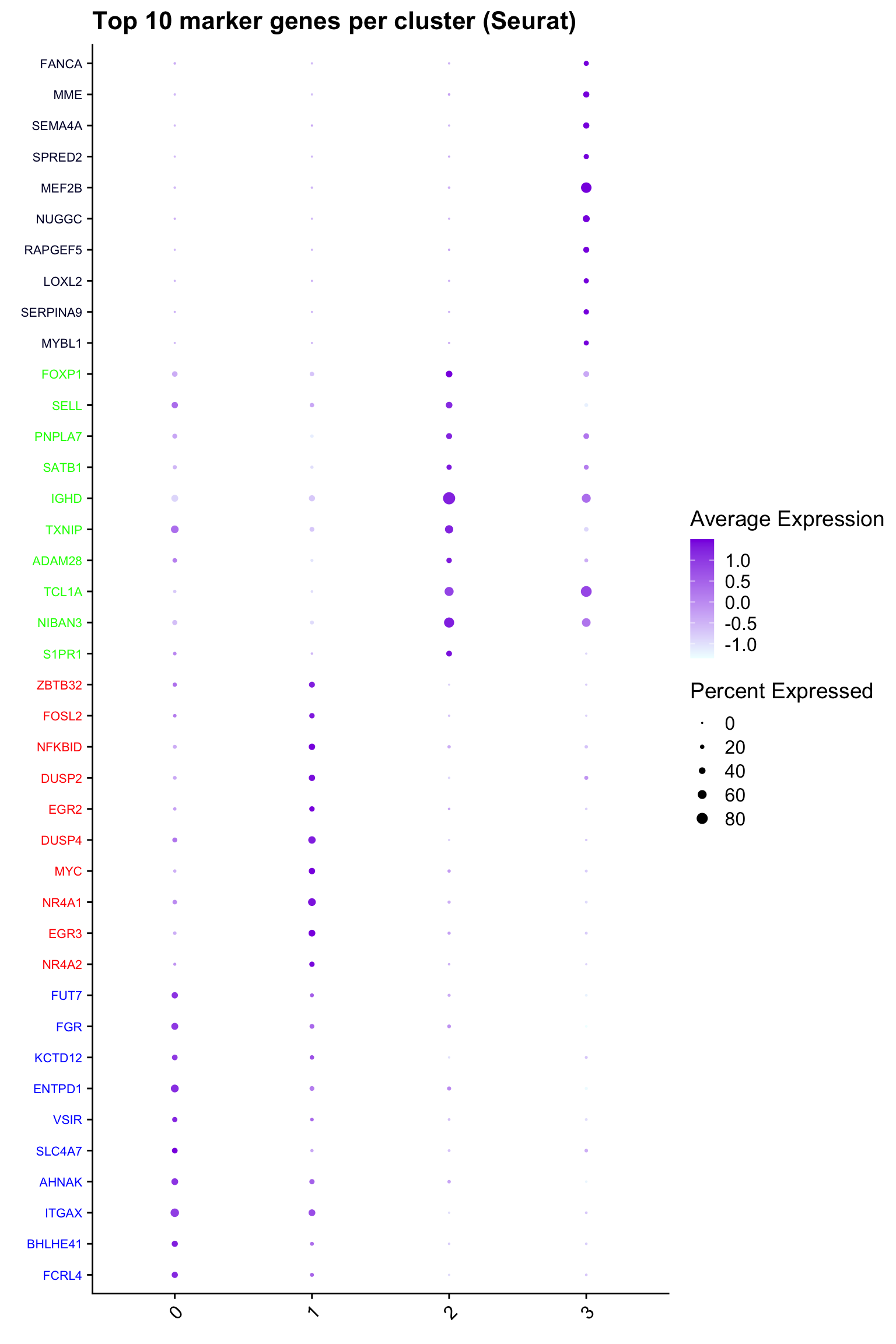
out_markers <- here("output",
"CSV",
paste(tissue,"_Marker_genes_Reclustered_Bcell_population.",opt_res, sep = ""))
dir.create(out_markers, recursive = TRUE, showWarnings = FALSE)
for (cl in unique(paed_sub.markers$cluster)) {
cluster_data <- paed_sub.markers %>% dplyr::filter(cluster == cl)
file_name <- here(out_markers, paste0("G000231_Neeland_",tissue, "_cluster_", cl, ".csv"))
write.csv(cluster_data, file = file_name)
}Save subclustered SEU object
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".Bcell_population.subclusters_without_DecontX.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Other Clusters (excluding subclusters)
idx <- which(Idents(seu_obj) %in% c("macro-monocyte-derived-or-interstitial", "macro-proliferating", "macro-lipid", "macro-alveolar", "macro-CCL", "CD4 T cells", "CD8 T cells", "NK-T cells", "proliferating T/NK", "cycling T cells"))
paed_sub <- seu_obj[,-idx]
paed_subAn object of class Seurat
17529 features across 5275 samples within 1 assay
Active assay: RNA (17529 features, 2000 variable features)
3 layers present: counts, data, scale.data
3 dimensional reductions calculated: pca, umap, umap.unintegratedpaed_sub$cell_labels_v2 <- Idents(paed_sub)# Visualize the clustering results
DimPlot(paed_sub, reduction = "umap", group.by = "cluster", label = TRUE, label.size = 2.5, repel = TRUE, raster = FALSE )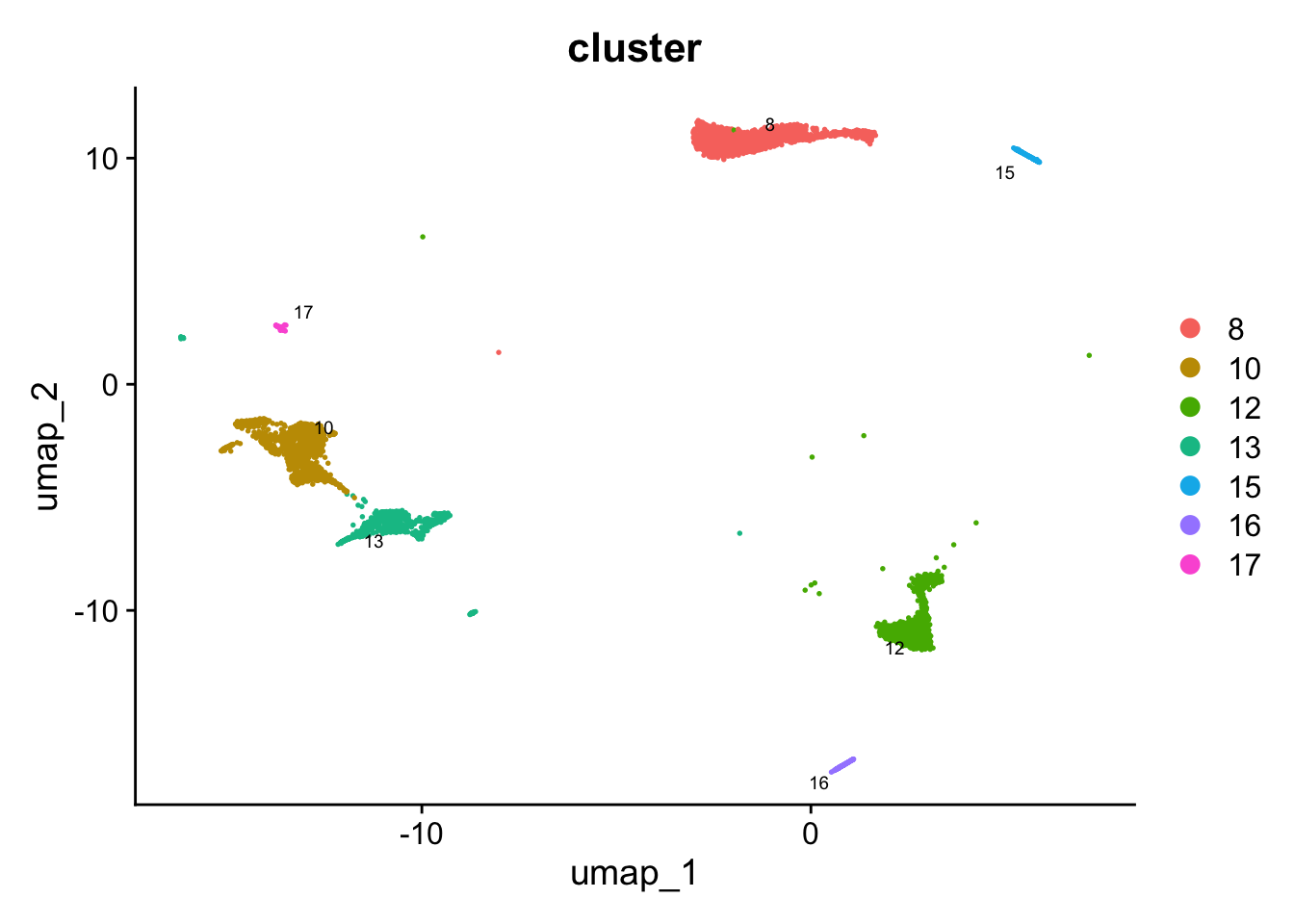
DimPlot(paed_sub, reduction = "umap", label = TRUE, label.size = 2.5, repel = TRUE, raster = FALSE )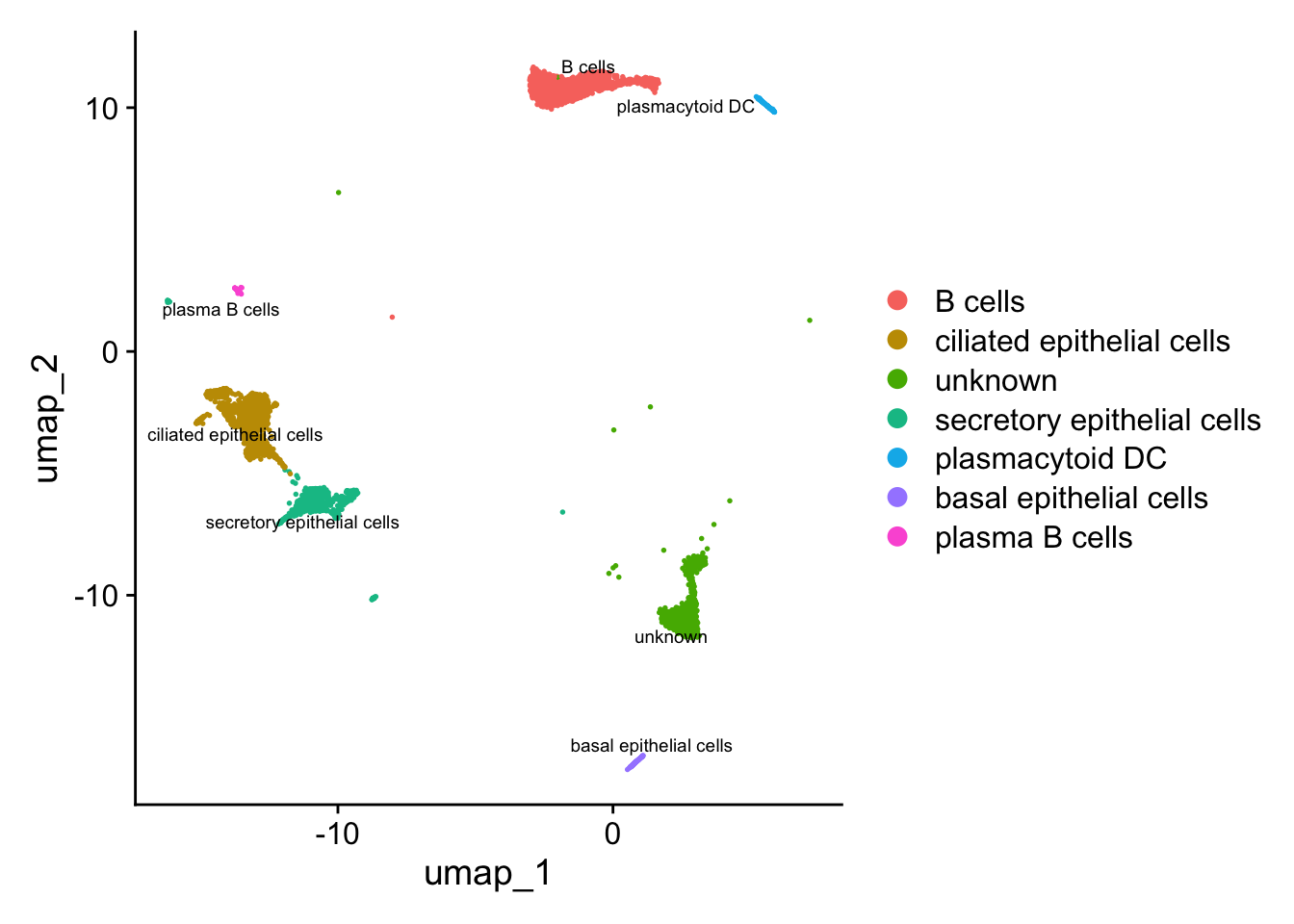
Save subclustered SEU object ( All other cells)
out2 <- here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue,
paste0("G000231_Neeland_",tissue,".all_other.subclusters.SEU.rds"))
#dir.create(out2)
if (!file.exists(out2)) {
saveRDS(paed_sub, file = out2)
}Merge seurat objects of subclusters
files <- list.files(here("output",
"RDS", "AllBatches_Subclustering_SEUs", tissue), pattern = "subclusters.SEU.rds",
full.names = TRUE)
seuLst <- lapply(files, function(f) readRDS(f))
seu <- merge(seuLst[[1]],
y = c(seuLst[[2]],
seuLst[[3]]))
seuExcluding contaminating labels
idx <- which(grepl("^contaminating", Idents(seu)))
seu <- seu[, -idx]merged <- seu %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
merged <- RunUMAP(merged, dims = 1:30, reduction = "pca", reduction.name = "umap.merged")
p4 <- DimPlot(merged, reduction = "umap.merged", group.by = "cell_labels_v2",raster = FALSE, repel = TRUE, label = TRUE, label.size = 4.5) + ggtitle(paste0(tissue, ": UMAP with annotations")) + NoLegend()
p4Save Final SEU object ( All cells)
merged@meta.data$donor_id <- sub("_\\d+$", "", merged@meta.data$Sample)
out3 <- here("output",
"RDS", "AllBatches_Final_Clusters_SEUs",
paste0("G000231_Neeland_",tissue,".final_clusters.SEU.rds"))
if (!file.exists(out3)) {
saveRDS(merged, file = out3)
}#metadata <- metadata %>%
# dplyr::select(-donor_id, -sample_id)
metadata <- merged@meta.data
batch_meta_subset <- batch_meta %>%
dplyr::select(sample_id, donor_id)
metadata <- metadata %>%
dplyr::left_join(batch_meta_subset, by = c("Sample" = "donor_id"))
merged@meta.data <- metadata
merged@meta.data$donor_id <- sub("_\\d+$", "", merged@meta.data$Sample)Session Info
sessioninfo::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.3.2 (2023-10-31)
os macOS 15.0.1
system aarch64, darwin20
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2024-10-11
pandoc 3.1.1 @ /Users/dixitgunjan/Desktop/RStudio.app/Contents/Resources/app/quarto/bin/tools/ (via rmarkdown)
─ Packages ───────────────────────────────────────────────────────────────────
package * version date (UTC) lib source
abind 1.4-5 2016-07-21 [1] CRAN (R 4.3.0)
AnnotationDbi * 1.64.1 2023-11-02 [1] Bioconductor
backports 1.4.1 2021-12-13 [1] CRAN (R 4.3.0)
Biobase * 2.62.0 2023-10-26 [1] Bioconductor
BiocGenerics * 0.48.1 2023-11-02 [1] Bioconductor
BiocManager 1.30.22 2023-08-08 [1] CRAN (R 4.3.0)
BiocStyle * 2.30.0 2023-10-26 [1] Bioconductor
Biostrings 2.70.2 2024-01-30 [1] Bioconductor 3.18 (R 4.3.2)
bit 4.0.5 2022-11-15 [1] CRAN (R 4.3.0)
bit64 4.0.5 2020-08-30 [1] CRAN (R 4.3.0)
bitops 1.0-7 2021-04-24 [1] CRAN (R 4.3.0)
blob 1.2.4 2023-03-17 [1] CRAN (R 4.3.0)
bslib 0.6.1 2023-11-28 [1] CRAN (R 4.3.1)
cachem 1.0.8 2023-05-01 [1] CRAN (R 4.3.0)
callr 3.7.5 2024-02-19 [1] CRAN (R 4.3.1)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 4.3.0)
checkmate 2.3.1 2023-12-04 [1] CRAN (R 4.3.1)
cli 3.6.2 2023-12-11 [1] CRAN (R 4.3.1)
cluster 2.1.6 2023-12-01 [1] CRAN (R 4.3.1)
clustree * 0.5.1 2023-11-05 [1] CRAN (R 4.3.1)
codetools 0.2-19 2023-02-01 [1] CRAN (R 4.3.2)
colorspace 2.1-0 2023-01-23 [1] CRAN (R 4.3.0)
cowplot 1.1.3 2024-01-22 [1] CRAN (R 4.3.1)
crayon 1.5.2 2022-09-29 [1] CRAN (R 4.3.0)
data.table * 1.15.0 2024-01-30 [1] CRAN (R 4.3.1)
DBI 1.2.2 2024-02-16 [1] CRAN (R 4.3.1)
DelayedArray 0.28.0 2023-11-06 [1] Bioconductor
deldir 2.0-2 2023-11-23 [1] CRAN (R 4.3.1)
digest 0.6.34 2024-01-11 [1] CRAN (R 4.3.1)
dotCall64 1.1-1 2023-11-28 [1] CRAN (R 4.3.1)
dplyr * 1.1.4 2023-11-17 [1] CRAN (R 4.3.1)
edgeR * 4.0.16 2024-02-20 [1] Bioconductor 3.18 (R 4.3.2)
ellipsis 0.3.2 2021-04-29 [1] CRAN (R 4.3.0)
evaluate 0.23 2023-11-01 [1] CRAN (R 4.3.1)
fansi 1.0.6 2023-12-08 [1] CRAN (R 4.3.1)
farver 2.1.1 2022-07-06 [1] CRAN (R 4.3.0)
fastDummies 1.7.3 2023-07-06 [1] CRAN (R 4.3.0)
fastmap 1.1.1 2023-02-24 [1] CRAN (R 4.3.0)
fitdistrplus 1.1-11 2023-04-25 [1] CRAN (R 4.3.0)
forcats * 1.0.0 2023-01-29 [1] CRAN (R 4.3.0)
fs 1.6.3 2023-07-20 [1] CRAN (R 4.3.0)
future 1.33.1 2023-12-22 [1] CRAN (R 4.3.1)
future.apply 1.11.1 2023-12-21 [1] CRAN (R 4.3.1)
generics 0.1.3 2022-07-05 [1] CRAN (R 4.3.0)
GenomeInfoDb 1.38.6 2024-02-10 [1] Bioconductor 3.18 (R 4.3.2)
GenomeInfoDbData 1.2.11 2024-02-27 [1] Bioconductor
GenomicRanges 1.54.1 2023-10-30 [1] Bioconductor
getPass 0.2-4 2023-12-10 [1] CRAN (R 4.3.1)
ggforce 0.4.2 2024-02-19 [1] CRAN (R 4.3.1)
ggplot2 * 3.5.0 2024-02-23 [1] CRAN (R 4.3.1)
ggraph * 2.1.0 2022-10-09 [1] CRAN (R 4.3.0)
ggrepel 0.9.5 2024-01-10 [1] CRAN (R 4.3.1)
ggridges 0.5.6 2024-01-23 [1] CRAN (R 4.3.1)
git2r 0.33.0 2023-11-26 [1] CRAN (R 4.3.1)
globals 0.16.2 2022-11-21 [1] CRAN (R 4.3.0)
glue * 1.7.0 2024-01-09 [1] CRAN (R 4.3.1)
goftest 1.2-3 2021-10-07 [1] CRAN (R 4.3.0)
graphlayouts 1.1.0 2024-01-19 [1] CRAN (R 4.3.1)
gridExtra 2.3 2017-09-09 [1] CRAN (R 4.3.0)
gtable 0.3.4 2023-08-21 [1] CRAN (R 4.3.0)
here * 1.0.1 2020-12-13 [1] CRAN (R 4.3.0)
highr 0.10 2022-12-22 [1] CRAN (R 4.3.0)
hms 1.1.3 2023-03-21 [1] CRAN (R 4.3.0)
htmltools 0.5.7 2023-11-03 [1] CRAN (R 4.3.1)
htmlwidgets 1.6.4 2023-12-06 [1] CRAN (R 4.3.1)
httpuv 1.6.14 2024-01-26 [1] CRAN (R 4.3.1)
httr 1.4.7 2023-08-15 [1] CRAN (R 4.3.0)
ica 1.0-3 2022-07-08 [1] CRAN (R 4.3.0)
igraph 2.0.2 2024-02-17 [1] CRAN (R 4.3.1)
IRanges * 2.36.0 2023-10-26 [1] Bioconductor
irlba 2.3.5.1 2022-10-03 [1] CRAN (R 4.3.2)
jquerylib 0.1.4 2021-04-26 [1] CRAN (R 4.3.0)
jsonlite 1.8.8 2023-12-04 [1] CRAN (R 4.3.1)
kableExtra * 1.4.0 2024-01-24 [1] CRAN (R 4.3.1)
KEGGREST 1.42.0 2023-10-26 [1] Bioconductor
KernSmooth 2.23-22 2023-07-10 [1] CRAN (R 4.3.2)
knitr 1.45 2023-10-30 [1] CRAN (R 4.3.1)
labeling 0.4.3 2023-08-29 [1] CRAN (R 4.3.0)
later 1.3.2 2023-12-06 [1] CRAN (R 4.3.1)
lattice 0.22-5 2023-10-24 [1] CRAN (R 4.3.1)
lazyeval 0.2.2 2019-03-15 [1] CRAN (R 4.3.0)
leiden 0.4.3.1 2023-11-17 [1] CRAN (R 4.3.1)
lifecycle 1.0.4 2023-11-07 [1] CRAN (R 4.3.1)
limma * 3.58.1 2023-11-02 [1] Bioconductor
listenv 0.9.1 2024-01-29 [1] CRAN (R 4.3.1)
lmtest 0.9-40 2022-03-21 [1] CRAN (R 4.3.0)
locfit 1.5-9.8 2023-06-11 [1] CRAN (R 4.3.0)
lubridate * 1.9.3 2023-09-27 [1] CRAN (R 4.3.1)
magrittr 2.0.3 2022-03-30 [1] CRAN (R 4.3.0)
MASS 7.3-60.0.1 2024-01-13 [1] CRAN (R 4.3.1)
Matrix 1.6-5 2024-01-11 [1] CRAN (R 4.3.1)
MatrixGenerics 1.14.0 2023-10-26 [1] Bioconductor
matrixStats 1.2.0 2023-12-11 [1] CRAN (R 4.3.1)
memoise 2.0.1 2021-11-26 [1] CRAN (R 4.3.0)
mime 0.12 2021-09-28 [1] CRAN (R 4.3.0)
miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.3.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 4.3.0)
nlme 3.1-164 2023-11-27 [1] CRAN (R 4.3.1)
org.Hs.eg.db * 3.18.0 2024-02-27 [1] Bioconductor
paletteer 1.6.0 2024-01-21 [1] CRAN (R 4.3.1)
parallelly 1.37.0 2024-02-14 [1] CRAN (R 4.3.1)
patchwork * 1.2.0 2024-01-08 [1] CRAN (R 4.3.1)
pbapply 1.7-2 2023-06-27 [1] CRAN (R 4.3.0)
pillar 1.9.0 2023-03-22 [1] CRAN (R 4.3.0)
pkgconfig 2.0.3 2019-09-22 [1] CRAN (R 4.3.0)
plotly 4.10.4 2024-01-13 [1] CRAN (R 4.3.1)
plyr 1.8.9 2023-10-02 [1] CRAN (R 4.3.1)
png 0.1-8 2022-11-29 [1] CRAN (R 4.3.0)
polyclip 1.10-6 2023-09-27 [1] CRAN (R 4.3.1)
presto 1.0.0 2024-02-27 [1] Github (immunogenomics/presto@31dc97f)
prismatic 1.1.1 2022-08-15 [1] CRAN (R 4.3.0)
processx 3.8.3 2023-12-10 [1] CRAN (R 4.3.1)
progressr 0.14.0 2023-08-10 [1] CRAN (R 4.3.0)
promises 1.2.1 2023-08-10 [1] CRAN (R 4.3.0)
ps 1.7.6 2024-01-18 [1] CRAN (R 4.3.1)
purrr * 1.0.2 2023-08-10 [1] CRAN (R 4.3.0)
R6 2.5.1 2021-08-19 [1] CRAN (R 4.3.0)
RANN 2.6.1 2019-01-08 [1] CRAN (R 4.3.0)
RColorBrewer * 1.1-3 2022-04-03 [1] CRAN (R 4.3.0)
Rcpp 1.0.12 2024-01-09 [1] CRAN (R 4.3.1)
RcppAnnoy 0.0.22 2024-01-23 [1] CRAN (R 4.3.1)
RcppHNSW 0.6.0 2024-02-04 [1] CRAN (R 4.3.1)
RCurl 1.98-1.14 2024-01-09 [1] CRAN (R 4.3.1)
readr * 2.1.5 2024-01-10 [1] CRAN (R 4.3.1)
readxl * 1.4.3 2023-07-06 [1] CRAN (R 4.3.0)
rematch2 2.1.2 2020-05-01 [1] CRAN (R 4.3.0)
reshape2 1.4.4 2020-04-09 [1] CRAN (R 4.3.0)
reticulate 1.35.0 2024-01-31 [1] CRAN (R 4.3.1)
rlang 1.1.3 2024-01-10 [1] CRAN (R 4.3.1)
rmarkdown 2.25 2023-09-18 [1] CRAN (R 4.3.1)
ROCR 1.0-11 2020-05-02 [1] CRAN (R 4.3.0)
rprojroot 2.0.4 2023-11-05 [1] CRAN (R 4.3.1)
RSpectra 0.16-1 2022-04-24 [1] CRAN (R 4.3.0)
RSQLite 2.3.5 2024-01-21 [1] CRAN (R 4.3.1)
rstudioapi 0.15.0 2023-07-07 [1] CRAN (R 4.3.0)
Rtsne 0.17 2023-12-07 [1] CRAN (R 4.3.1)
S4Arrays 1.2.0 2023-10-26 [1] Bioconductor
S4Vectors * 0.40.2 2023-11-25 [1] Bioconductor 3.18 (R 4.3.2)
sass 0.4.8 2023-12-06 [1] CRAN (R 4.3.1)
scales 1.3.0 2023-11-28 [1] CRAN (R 4.3.1)
scattermore 1.2 2023-06-12 [1] CRAN (R 4.3.0)
sctransform 0.4.1 2023-10-19 [1] CRAN (R 4.3.1)
sessioninfo 1.2.2 2021-12-06 [1] CRAN (R 4.3.0)
Seurat * 5.0.1.9009 2024-02-28 [1] Github (satijalab/seurat@6a3ef5e)
SeuratObject * 5.0.1 2023-11-17 [1] CRAN (R 4.3.1)
shiny 1.8.0 2023-11-17 [1] CRAN (R 4.3.1)
SingleCellExperiment 1.24.0 2023-11-06 [1] Bioconductor
sp * 2.1-3 2024-01-30 [1] CRAN (R 4.3.1)
spam 2.10-0 2023-10-23 [1] CRAN (R 4.3.1)
SparseArray 1.2.4 2024-02-10 [1] Bioconductor 3.18 (R 4.3.2)
spatstat.data 3.0-4 2024-01-15 [1] CRAN (R 4.3.1)
spatstat.explore 3.2-6 2024-02-01 [1] CRAN (R 4.3.1)
spatstat.geom 3.2-8 2024-01-26 [1] CRAN (R 4.3.1)
spatstat.random 3.2-2 2023-11-29 [1] CRAN (R 4.3.1)
spatstat.sparse 3.0-3 2023-10-24 [1] CRAN (R 4.3.1)
spatstat.utils 3.0-4 2023-10-24 [1] CRAN (R 4.3.1)
speckle * 1.2.0 2023-10-26 [1] Bioconductor
statmod 1.5.0 2023-01-06 [1] CRAN (R 4.3.0)
stringi 1.8.3 2023-12-11 [1] CRAN (R 4.3.1)
stringr * 1.5.1 2023-11-14 [1] CRAN (R 4.3.1)
SummarizedExperiment 1.32.0 2023-11-06 [1] Bioconductor
survival 3.5-8 2024-02-14 [1] CRAN (R 4.3.1)
svglite 2.1.3 2023-12-08 [1] CRAN (R 4.3.1)
systemfonts 1.0.5 2023-10-09 [1] CRAN (R 4.3.1)
tensor 1.5 2012-05-05 [1] CRAN (R 4.3.0)
tibble * 3.2.1 2023-03-20 [1] CRAN (R 4.3.0)
tidygraph 1.3.1 2024-01-30 [1] CRAN (R 4.3.1)
tidyr * 1.3.1 2024-01-24 [1] CRAN (R 4.3.1)
tidyselect 1.2.0 2022-10-10 [1] CRAN (R 4.3.0)
tidyverse * 2.0.0 2023-02-22 [1] CRAN (R 4.3.0)
timechange 0.3.0 2024-01-18 [1] CRAN (R 4.3.1)
tweenr 2.0.3 2024-02-26 [1] CRAN (R 4.3.1)
tzdb 0.4.0 2023-05-12 [1] CRAN (R 4.3.0)
utf8 1.2.4 2023-10-22 [1] CRAN (R 4.3.1)
uwot 0.1.16 2023-06-29 [1] CRAN (R 4.3.0)
vctrs 0.6.5 2023-12-01 [1] CRAN (R 4.3.1)
viridis 0.6.5 2024-01-29 [1] CRAN (R 4.3.1)
viridisLite 0.4.2 2023-05-02 [1] CRAN (R 4.3.0)
whisker 0.4.1 2022-12-05 [1] CRAN (R 4.3.0)
withr 3.0.0 2024-01-16 [1] CRAN (R 4.3.1)
workflowr * 1.7.1 2023-08-23 [1] CRAN (R 4.3.0)
xfun 0.42 2024-02-08 [1] CRAN (R 4.3.1)
xml2 1.3.6 2023-12-04 [1] CRAN (R 4.3.1)
xtable 1.8-4 2019-04-21 [1] CRAN (R 4.3.0)
XVector 0.42.0 2023-10-26 [1] Bioconductor
yaml 2.3.8 2023-12-11 [1] CRAN (R 4.3.1)
zlibbioc 1.48.0 2023-10-26 [1] Bioconductor
zoo 1.8-12 2023-04-13 [1] CRAN (R 4.3.0)
[1] /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/library
──────────────────────────────────────────────────────────────────────────────
sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS 15.0.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Australia/Melbourne
tzcode source: internal
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] readxl_1.4.3 org.Hs.eg.db_3.18.0 AnnotationDbi_1.64.1
[4] IRanges_2.36.0 S4Vectors_0.40.2 Biobase_2.62.0
[7] BiocGenerics_0.48.1 speckle_1.2.0 edgeR_4.0.16
[10] limma_3.58.1 patchwork_1.2.0 data.table_1.15.0
[13] RColorBrewer_1.1-3 kableExtra_1.4.0 clustree_0.5.1
[16] ggraph_2.1.0 Seurat_5.0.1.9009 SeuratObject_5.0.1
[19] sp_2.1-3 glue_1.7.0 here_1.0.1
[22] lubridate_1.9.3 forcats_1.0.0 stringr_1.5.1
[25] dplyr_1.1.4 purrr_1.0.2 readr_2.1.5
[28] tidyr_1.3.1 tibble_3.2.1 ggplot2_3.5.0
[31] tidyverse_2.0.0 BiocStyle_2.30.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] fs_1.6.3 matrixStats_1.2.0
[3] spatstat.sparse_3.0-3 bitops_1.0-7
[5] httr_1.4.7 tools_4.3.2
[7] sctransform_0.4.1 backports_1.4.1
[9] utf8_1.2.4 R6_2.5.1
[11] lazyeval_0.2.2 uwot_0.1.16
[13] withr_3.0.0 gridExtra_2.3
[15] progressr_0.14.0 cli_3.6.2
[17] spatstat.explore_3.2-6 fastDummies_1.7.3
[19] prismatic_1.1.1 labeling_0.4.3
[21] sass_0.4.8 spatstat.data_3.0-4
[23] ggridges_0.5.6 pbapply_1.7-2
[25] systemfonts_1.0.5 svglite_2.1.3
[27] sessioninfo_1.2.2 parallelly_1.37.0
[29] rstudioapi_0.15.0 RSQLite_2.3.5
[31] generics_0.1.3 ica_1.0-3
[33] spatstat.random_3.2-2 Matrix_1.6-5
[35] fansi_1.0.6 abind_1.4-5
[37] lifecycle_1.0.4 whisker_0.4.1
[39] yaml_2.3.8 SummarizedExperiment_1.32.0
[41] SparseArray_1.2.4 Rtsne_0.17
[43] paletteer_1.6.0 grid_4.3.2
[45] blob_1.2.4 promises_1.2.1
[47] crayon_1.5.2 miniUI_0.1.1.1
[49] lattice_0.22-5 cowplot_1.1.3
[51] KEGGREST_1.42.0 pillar_1.9.0
[53] knitr_1.45 GenomicRanges_1.54.1
[55] future.apply_1.11.1 codetools_0.2-19
[57] leiden_0.4.3.1 getPass_0.2-4
[59] vctrs_0.6.5 png_0.1-8
[61] spam_2.10-0 cellranger_1.1.0
[63] gtable_0.3.4 rematch2_2.1.2
[65] cachem_1.0.8 xfun_0.42
[67] S4Arrays_1.2.0 mime_0.12
[69] tidygraph_1.3.1 survival_3.5-8
[71] SingleCellExperiment_1.24.0 statmod_1.5.0
[73] ellipsis_0.3.2 fitdistrplus_1.1-11
[75] ROCR_1.0-11 nlme_3.1-164
[77] bit64_4.0.5 RcppAnnoy_0.0.22
[79] GenomeInfoDb_1.38.6 rprojroot_2.0.4
[81] bslib_0.6.1 irlba_2.3.5.1
[83] KernSmooth_2.23-22 colorspace_2.1-0
[85] DBI_1.2.2 tidyselect_1.2.0
[87] processx_3.8.3 bit_4.0.5
[89] compiler_4.3.2 git2r_0.33.0
[91] xml2_1.3.6 DelayedArray_0.28.0
[93] plotly_4.10.4 checkmate_2.3.1
[95] scales_1.3.0 lmtest_0.9-40
[97] callr_3.7.5 digest_0.6.34
[99] goftest_1.2-3 spatstat.utils_3.0-4
[101] presto_1.0.0 rmarkdown_2.25
[103] XVector_0.42.0 htmltools_0.5.7
[105] pkgconfig_2.0.3 MatrixGenerics_1.14.0
[107] highr_0.10 fastmap_1.1.1
[109] rlang_1.1.3 htmlwidgets_1.6.4
[111] shiny_1.8.0 farver_2.1.1
[113] jquerylib_0.1.4 zoo_1.8-12
[115] jsonlite_1.8.8 RCurl_1.98-1.14
[117] magrittr_2.0.3 GenomeInfoDbData_1.2.11
[119] dotCall64_1.1-1 munsell_0.5.0
[121] Rcpp_1.0.12 viridis_0.6.5
[123] reticulate_1.35.0 stringi_1.8.3
[125] zlibbioc_1.48.0 MASS_7.3-60.0.1
[127] plyr_1.8.9 parallel_4.3.2
[129] listenv_0.9.1 ggrepel_0.9.5
[131] deldir_2.0-2 Biostrings_2.70.2
[133] graphlayouts_1.1.0 splines_4.3.2
[135] tensor_1.5 hms_1.1.3
[137] locfit_1.5-9.8 ps_1.7.6
[139] igraph_2.0.2 spatstat.geom_3.2-8
[141] RcppHNSW_0.6.0 reshape2_1.4.4
[143] evaluate_0.23 BiocManager_1.30.22
[145] tzdb_0.4.0 tweenr_2.0.3
[147] httpuv_1.6.14 RANN_2.6.1
[149] polyclip_1.10-6 future_1.33.1
[151] scattermore_1.2 ggforce_0.4.2
[153] xtable_1.8-4 RSpectra_0.16-1
[155] later_1.3.2 viridisLite_0.4.2
[157] memoise_2.0.1 cluster_2.1.6
[159] timechange_0.3.0 globals_0.16.2