 Microbiome
Microbiome
McConville
Lab
BMDM Screen QC
Last updated: 2025-03-24
Checks: 6 1
Knit directory:
McConville_Lab_Microbiome_PMC255A/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R
Markdown file created these results, you’ll want to first commit it to
the Git repo. If you’re still working on the analysis, you can ignore
this warning. When you’re finished, you can run
wflow_publish to commit the R Markdown file and build the
HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20250117) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 3a65c0b. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: data/raw_data/
Untracked files:
Untracked: analysis/PMC255A_ScreenQC_v4.Rmd
Untracked: analysis/prelim/
Unstaged changes:
Modified: analysis/PMC255A_ScreenComparison.Rmd
Deleted: analysis/PMC255A_ScreenQC_v3.Rmd
Modified: analysis/_site.yml
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with
wflow_publish() to start tracking its development.
Aim
To examine the polarisation potential of BDBM cells from M0 to M1 or M2 in the presence of compounds and different concentrations of polarisation activator.
Researcher
Ada Koo, McConville Lab, Bedoui Lab
Readout
Image analysis of BMDM cells stained with DAPI, ARG, INOS, and BODIPY (CX7 Pro LZR, 9 fields0 @ 20x)
Image analysis software
CellProfiler 4.1.3
Required R packages
data.table, DT, reshape2, tidyverse, viridis, patchwork
# load the required packages
library(data.table)
library(readxl)
library(DT)
library(reshape2)
library(viridis)
library(patchwork)
library(tidyverse)
library(magick)
# fix ggplotly axis labels
layout_ggplotly <- function(gg, x = -0.02, y = -0.08){
# The 1 and 2 goes into the list that contains the options for the x and y axis labels respectively
gg[['x']][['layout']][['annotations']][[1]][['y']] <- x
gg[['x']][['layout']][['annotations']][[2]][['x']] <- y
gg
}
# set the file prefix
prefix <- "KW_PMC255A"
# turn off scientific notation
options(scipen=999)Screen details
Screen dates (yyyy-mm-dd)
2024-12-04
Cell lines
BMDM M0
Conditions
unstimulated (Unstim_M0)
5 ng/mL IL4 + 5 ng/mL IL13 (Lo_M2)
10 ng/mL IL4 + 10 ng/mL IL13 (Hi_M2)
3 ng/mL IFNg + 3 ng/mL LPS
(Lo_M1)
25 ng/mL IFNg + 25 ng/mL LPS (Hi_M1)
25 ng/mL IFNg
(Potential_M1)
Screen outline
Library: Custom MCE library
Library plates: 4
Cell plates
per library plate: 24
Controls
Per well data is normalised to Stimulated DMSO on a per plate basis.
Known technical issues
None.
Plate layout
get_image_dimensions <- function(path) {
img_info <- magick::image_info(magick::image_read(path))
setNames(c(img_info$width, img_info$height), c("Width", "Height"))
}
# Example usage
path <- file.path("data", paste0(prefix, "_platemap.png"))
img_dims <- get_image_dimensions(path)
# Generate the HTML image with scaled dimensions
htmltools::img(
src = knitr::image_uri(path),
style = sprintf("height:%.0fpx;width:%.0fpx;", img_dims["Height"] / 2, img_dims["Width"] / 2)
)
Data cleaning
The raw data was read into R Studio.
data_raw_obj <- list.files(path = paste0("data/raw_data/PerObject/"), # list all .csv files
pattern = "*.csv",
full.names = T) %>%
set_names(nm = basename(.)) %>%
map_dfr(., fread, .id = "FileName") %>% # Add file name as a column
mutate(FileName = tools::file_path_sans_ext(FileName)) %>% # remove .csv extensions
as.data.frame() %>%
mutate(Barcode = FileName,
Barcode = gsub("PerObject_|_Cytoplasm", "", Barcode),
Barcode = gsub("Rep", "Plate", Barcode),
Barcode = gsub("Unsure", "Potential", Barcode)) %>%
select(Barcode,
PlateID = matches("^Metadata_PlateID", ignore.case = FALSE)[1],
WellID = matches("^Metadata_WellID", ignore.case = FALSE)[1],
Field = matches("^Metadata_Field", ignore.case = FALSE)[1],
Bodipy_spots_count = Children_BODIPY_spots_Count,
Intensity_IntegratedIntensity_BODIPY = matches("^Intensity_IntegratedIntensity_BODIPY", ignore.case = FALSE)[1],
Intensity_MeanIntensity_BODIPY = matches("^Intensity_MeanIntensity_BODIPY", ignore.case = FALSE)[1],
Intensity_StdIntensity_BODIPY = matches("^Intensity_StdIntensity_BODIPY", ignore.case = FALSE)[1],
contains(c("ARG", "INOS")))
# data_raw_obj %>% #H01
# filter(grepl("A17", WellID) & grepl("Unstim_M0_Plate4", Barcode)) %>%
# select(Field, contains(c("MeanIntensity"))) %>%
# group_by(Field) %>%
# summarise(across(contains(c("MeanIntensity")), lst(sum)))Data processing
## Annotation
Well annotations were added to the Per Object data.
# read in annotation files
plate_info <- read_csv(paste0("data/KW_PMC255A_barcodes.csv"), show_col_types = FALSE)
annotation <- read_csv(paste0("data/annot/", prefix, "_annotation.csv"), show_col_types = FALSE)
# annotate data
data_annot_obj <- data_raw_obj %>%
left_join(plate_info, ., by = c("Barcode", "PlateID")) %>%
left_join(annotation, ., by = c("WellID", "VCFG_Plate_ID" = "Replicate")) %>%
select(-PlateID2) %>%
# fix concentration, units and labels for each plate
mutate(Concentration = case_when(Compound_Name == "IL4+IL13" ~ 50,
Compound_Name == "LPS+INFg" ~ 50,
TRUE ~ Concentration),
Units = case_when(grepl("IL4\\+IL13|LPS\\+INFg", Compound_Name) ~ "ng/mL",
TRUE ~ Units),
Stimulation = case_when(grepl("01", WellID) ~ "Unstimulated", TRUE ~ "Stimulated"))
# write out image annot file
write_excel_csv(data_annot_obj, "output/data_annot_obj.csv")# clean up annotation files
annotation <- read_csv(paste0("data/annot/", prefix, "_annotation.csv"), show_col_types = FALSE)
plate_info <- read_csv(paste0("data/KW_PMC255A_barcodes.csv"), show_col_types = FALSE)
# replace non-alphanumeric characters in compound names
library(tidyverse)
library(data.table)
library(stringi)
annot_clean <- annotation %>%
mutate(Compound_Name_Clean = gsub(" ", "_", Compound_Name),
Compound_Name_Clean = stri_trans_general(Compound_Name_Clean, "Latin-ASCII"),
Compound_Name_Clean = gsub("α", "alpha", Compound_Name_Clean),
Compound_Name_Clean = gsub("β", "beta", Compound_Name_Clean),
Compound_Name_Clean = gsub("γ", "gamma", Compound_Name_Clean),
Compound_Name_Clean = gsub("κ", "kappa", Compound_Name_Clean),
Compound_Name_Clean = gsub("Δ|δ", "delta", Compound_Name_Clean),
Compound_Name_Clean = gsub("Θ|θ", "theta", Compound_Name_Clean),
Compound_Name_Clean = gsub("ι", "iota", Compound_Name_Clean),
Compound_Name_Clean = gsub("Λ|λ", "lambda", Compound_Name_Clean),
Compound_Name_Clean = gsub("μ", "mu", Compound_Name_Clean),
Compound_Name_Clean = gsub("ν", "nu", Compound_Name_Clean),
Compound_Name_Clean = gsub("Σ|σ", "sigma", Compound_Name_Clean),
Compound_Name_Clean = gsub("Φ|φ", "phi", Compound_Name_Clean),
Compound_Name_Clean = gsub("Π|π", "pi", Compound_Name_Clean),
Compound_Name_Clean = gsub("Ψ|ψ", "psi", Compound_Name_Clean),
Compound_Name_Clean = gsub("Ω|ω", "omega", Compound_Name_Clean),
Compound_Name_Clean = gsub("ρ", "rho", Compound_Name_Clean),
Compound_Name_Clean = gsub("[^[:alnum:]]", "", Compound_Name_Clean))# Find rows containing non-English characters in the text_column
non_english_rows <- grep("[^[:alnum:]_-]", annot_clean$Compound_ID_Clean, value = FALSE)
# Subset dataframe to rows with non-English characters
df_non_english <- annot_clean[non_english_rows, ]
View(df_non_english)annot_clean_complete <- annot_clean %>%
inner_join(., plate_info, by = c("VCFG_Plate_ID" = "Replicate")) %>%
select(PlateID, VCFG_Plate_ID, Barcode, WellID, QCL_Sample_Number, Compound_Name, Compound_Name_Clean, Concentration, Units) %>%
distinct()
write_csv(annot_clean_complete, paste0("output/", prefix, "_image_annotation.csv"))data_annot_obj <- read_csv("output/data_annot_obj.csv")Thresholding
Histograms below shows the count of percentage positive cells for ARG and INOS staining based on the negative control (Unstimulated DMSO). Dotted red line indicates the cut off used for thresholding positive percentage cells in the Unstimulated DMSO wells across all plates.
Thresholds for percentage positive cells were set globally based on
the following limits:
* ARG positive threshold (MeanIntensity >=
mean_intensity_ARG of DMSO + 1.5 x std_dev_ARG)
* INOS positive
threshold (MeanIntensity >= mean_intensity_INOS of DMSO + 1.25 x
std_dev_INOS)
* BODIPY positive (>= 2 spot count)
# create function to generate density plot
gendensityplot <- function(vars, limits){
# for labelling purposes
data_annot_obj$VCFG_Plate_ID <- paste("VCFG_Plate_ID:", data_annot_obj$VCFG_Plate_ID)
# filter data and add polarity description
data_annot_obj_filt <- data_annot_obj %>%
filter((!grepl("Media", Compound_Name)) & grepl("01", WellID))
# calculate the median and standard deviation per plate
stats_perplate <- ungroup(data_annot_obj_filt) %>%
filter(Compound_Name == "DMSO") %>% # use untreated instead of treated DMSO
group_by(VCFG_Plate_ID) %>%
summarise(
mean_intensity = mean(.data[[vars]], na.rm = TRUE),
std_dev = sd(.data[[vars]], na.rm = TRUE),
threshold_low = mean_intensity - limits * std_dev,
threshold_high = mean_intensity + ifelse(VCFG_Plate_ID == "Plate4", 0.75, limits) * std_dev,
lim = limits)
data_annot_obj$VCFG_Plate_ID <- paste("VCFG_Plate_ID:", data_annot_obj$VCFG_Plate_ID)
# plot density plot
ggplot(data_annot_obj_filt, aes(x = .data[[vars]], fill = Compound_Name, color = Compound_Name)) +
geom_density(alpha = 0.4) +
geom_vline(data = stats_perplate, aes(xintercept = threshold_high), color = "black", linetype = "dashed", size = 0.5) +
facet_wrap(~VCFG_Plate_ID+Compound_Name, scales = "free", ncol = 6) +
labs(title = vars, x = vars, y = "Density") +
theme_bw() +
theme(
plot.title = element_text(size = 14),
axis.text.y = element_text(size = 11),
axis.text.x = element_text(size = 11),
legend.title = element_text(size = 12),
legend.text = element_text(size = 11))
}
# test different limits to determine final threshold cut off for percentage positive cells
(gendensityplot("Intensity_MeanIntensity_ARG", 1.5) /
gendensityplot("Intensity_MeanIntensity_INOS", 1.25)) /
plot_layout(guides = "collect")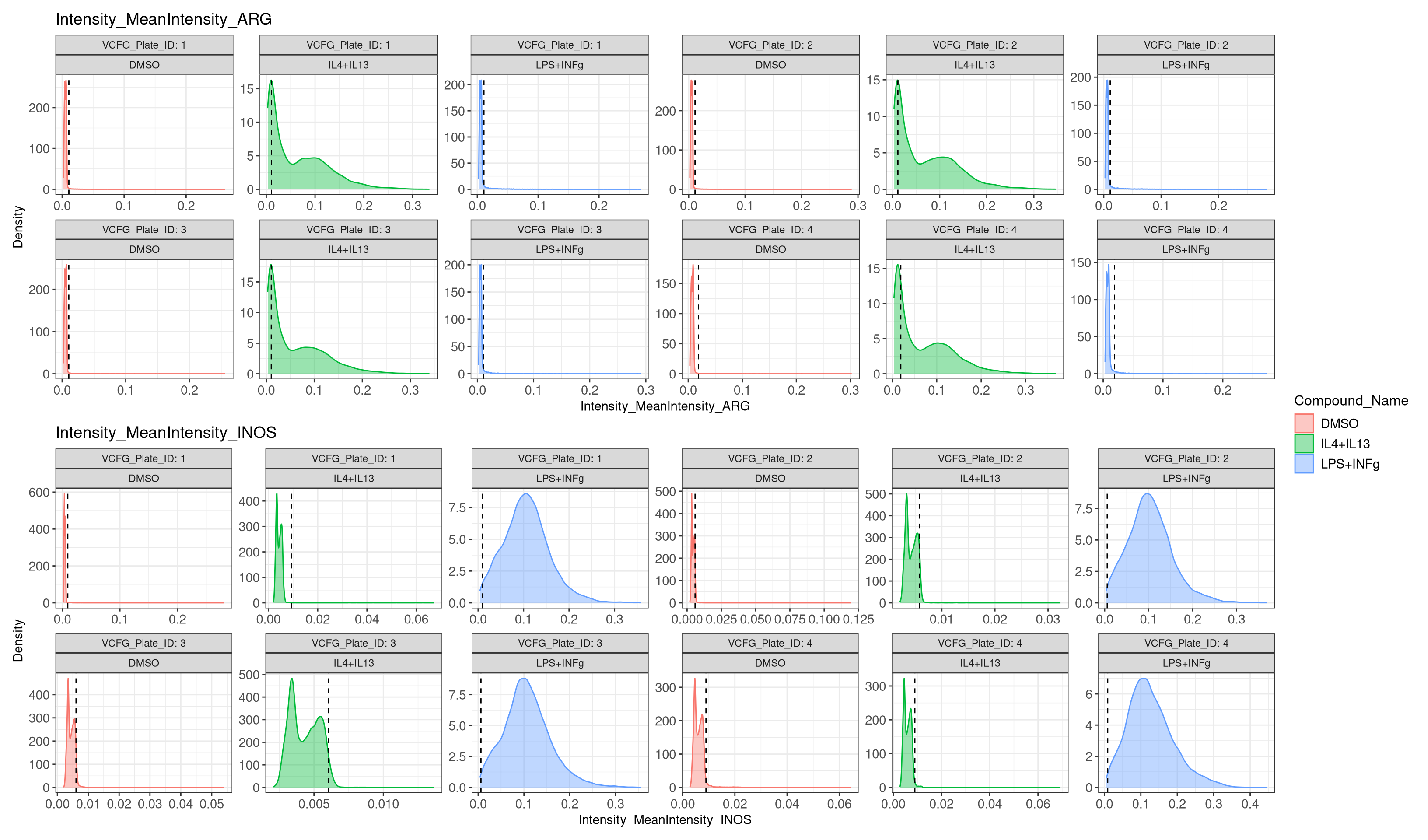
# create function to generate density plot
gendensityplot <- function(vars, limits){
# filter data and add polarity description
data_annot_obj_filt <- data_annot_obj %>%
filter((!grepl("Media", Compound_Name)) & grepl("01", WellID))
# calculate the median and standard deviation per plate
stats_perplate <- ungroup(data_annot_obj_filt) %>%
filter(Compound_Name == "DMSO") %>% # use untreated instead of treated DMSO
group_by(Barcode) %>%
summarise(
mean_intensity = mean(.data[[vars]], na.rm = TRUE),
std_dev = sd(.data[[vars]], na.rm = TRUE),
threshold_low = mean_intensity - limits * std_dev,
threshold_high = mean_intensity + limits * std_dev,
lim = limits)
# plot density plot
ggplot(data_annot_obj_filt, aes(x = .data[[vars]], fill = Compound_Name, color = Compound_Name)) +
geom_density(alpha = 0.4) +
geom_vline(data = stats_perplate, aes(xintercept = threshold_high), color = "black", linetype = "dashed", size = 0.5) +
facet_wrap(~Barcode+Compound_Name, scales = "free", ncol = 12) +
labs(title = vars, x = vars, y = "Density") +
theme_bw() +
theme(
legend.position = 'top',
plot.title = element_text(size = 14),
axis.text.y = element_text(size = 11),
axis.text.x = element_text(size = 11),
legend.title = element_text(size = 12),
legend.text = element_text(size = 11))
}
# test different limits to determine final threshold cut off for percentage positive cells
plot1 <- gendensityplot("Intensity_MeanIntensity_ARG", 1.5)
plot2 <- gendensityplot("Intensity_MeanIntensity_INOS", 1.25)
# save the plot as a PDF
pdf(paste0("output/", prefix, "_DensityPlots.pdf"), width = 25, height = 13)
print(plot1)
print(plot2)
dev.off()Aggregation
The per-cell data was aggregated into per-well data (total cell
counts and % positive cells in each channel).
# generate limits
neg_limits <- data_annot_obj %>%
group_by(Barcode) %>%
filter(grepl("01", WellID) & Compound_Name == "DMSO") %>%
summarise(arg_pos_limit = (mean(Intensity_MeanIntensity_ARG) + (1.5*sd(Intensity_MeanIntensity_ARG))),
inos_pos_limit = (mean(Intensity_MeanIntensity_INOS) + (1.25*sd(Intensity_MeanIntensity_INOS))),
bodipy_pos_limit = 2)
data_perwell <- data_annot_obj %>%
left_join(., neg_limits, by = c("Barcode")) %>%
mutate(
ARG_label = case_when(
Intensity_MeanIntensity_ARG >= arg_pos_limit ~ "ARG_positive", TRUE ~ "ARG_negative"),
INOS_label = case_when(
Intensity_MeanIntensity_INOS >= inos_pos_limit ~ "INOS_positive", TRUE ~ "INOS_negative"),
`INOS+BODIPY_label` = case_when((Intensity_MeanIntensity_INOS >= inos_pos_limit & Bodipy_spots_count >=
bodipy_pos_limit) ~ "INOS+BODIPY_positive", TRUE ~ "INOS+BODIPY_negative"),
`INOS-BODIPY_label` = case_when((Intensity_MeanIntensity_INOS >= inos_pos_limit & Bodipy_spots_count <
bodipy_pos_limit) ~ "INOS-BODIPY_positive", TRUE ~ "INOS-BODIPY_negative"),
`BODIPY_label` = case_when(Bodipy_spots_count >=
bodipy_pos_limit ~ "BODIPY_positive", TRUE ~ "BODIPY_negative")) %>%
mutate(Condition_Name = case_when(grepl("Unstim_M0", Barcode) ~ "Unstimulated",
grepl("Potential_M1", Barcode) ~ "25 ng/mL INFg",
grepl("Lo_M1", Barcode) ~ "3 ng/mL INFg + 3 ng/mL LPS",
grepl("Hi_M1", Barcode) ~ "25 ng/mL INFg + 25 ng/mL LPS",
grepl("Lo_M2", Barcode) ~ "5 ng/mL IL4 + 5 ng/mL IL13",
grepl("Hi_M2", Barcode) ~ "10 ng/mL IL4 + 10 ng/mL IL13"),
Phenotype = case_when(grepl("Unstim_M0", Barcode) ~ "M0",
grepl("Potential_M1", Barcode) ~ "M1",
grepl("Lo_M1", Barcode) ~ "M1",
grepl("Hi_M1", Barcode) ~ "M1",
grepl("Lo_M2", Barcode) ~ "M2",
grepl("Hi_M2", Barcode) ~ "M2")) %>%
group_by(Barcode, PlateID, Description, WellID, QCL_Sample_Number, Compound_Name, Concentration, Units, VCFG_Plate_ID, Target, Pathway, Condition_Name, Phenotype, Stimulation) %>%
summarise(Total_DAPI_Count = n(),
# Total_MeanIntensity_ARG = mean(Intensity_MeanIntensity_ARG),
# Total_MeanIntensity_INOS = mean(Intensity_MeanIntensity_INOS),
# Total_StdIntensity_BODIPY = mean(Intensity_StdIntensity_BODIPY),
Positive_Perc_ARG = sum(ARG_label == "ARG_positive")/n() * 100,
Positive_Perc_INOS = sum(INOS_label == "INOS_positive")/n() * 100,
`Positive_Perc_INOS+BODIPY` = sum(`INOS+BODIPY_label` == "INOS+BODIPY_positive")/n() * 100,
`Positive_Perc_INOS-BODIPY` = sum(`INOS-BODIPY_label` == "INOS-BODIPY_positive")/n() * 100,
`Positive_Perc_BODIPY` = sum(`BODIPY_label` == "BODIPY_positive")/n() * 100,
`CellCount_BODIPY` =sum(`BODIPY_label` == "BODIPY_positive"),
`SpotCount_BODIPY` = sum(`Bodipy_spots_count`),
`MeanSpotCount_PerBODIPYPosCell` = (`SpotCount_BODIPY`/`CellCount_BODIPY`),
`MeanSpotCount_PerCell` = `SpotCount_BODIPY`/n()) %>%
select(-`CellCount_BODIPY`, -`SpotCount_BODIPY`)
# create a df for controls
data_perwell_controls <- data_perwell %>%
filter(grepl('Control', Description))
# write out the data
write_csv(data_perwell_controls, paste0("output/", prefix, "_data_perwell_controls.csv"))
write_csv(data_perwell, paste0("output/", prefix, "_data_perwell.csv"))Normalisation
The raw values were normalised by fold changing to the median of the negative control (DMSO) wells within each plate.
# melt the dataframe
id <- colnames(data_perwell[1:14])
measure <- colnames(data_perwell[15:22])
data_perwell_melt <- data_perwell %>%
select(1:14, measure) %>%
melt(id.vars = id, measure.vars = measure) %>%
select(everything(), Raw = value, Feature = variable) %>%
arrange(Barcode, Feature, WellID)
# normalise dataframe
data_norm_melt <- ungroup(data_perwell_melt) %>%
group_by(Barcode, Feature) %>%
mutate(Raw = case_when(Raw == 0 ~ 0.00001, TRUE ~ Raw)) %>%
mutate(
Compound_Name = case_when(grepl("01", WellID) ~ paste0(Compound_Name, "_Unstim"), TRUE ~ Compound_Name),
NormToUnstimDMSO = Raw / median(Raw[Compound_Name == "DMSO_Unstim"], na.rm = TRUE),
NormToStimDMSO = Raw / median(Raw[Compound_Name == "DMSO"], na.rm = TRUE)) %>%
left_join(., data_perwell_melt, by = c("Barcode", "PlateID", "Description", "WellID", "QCL_Sample_Number", "Compound_Name", "Concentration", "Units", "VCFG_Plate_ID", "Target", "Pathway", "Condition_Name", "Phenotype", "Stimulation", "Raw", "Feature")) %>%
ungroup() %>%
mutate(Description = case_when(grepl("Unstim_M0_Plate4", Barcode) & grepl("A17", WellID) & grepl("INOS", Feature) ~ "Excluded", TRUE ~ Description))
# unmelt the dataframe
data_norm <- data_norm_melt %>%
pivot_wider(names_from = Feature, values_from = c(Raw, NormToUnstimDMSO, NormToStimDMSO), names_glue = "{.value}_{Feature}")
write_excel_csv(data_norm, paste0("output/", prefix, "_data_norm.csv"))
write_excel_csv(data_norm_melt, paste0("output/", prefix, "_data_norm_melt.csv"))# read in data
# data_norm_melt <- read.csv(paste0("output/", prefix, "_data_norm_melt.csv"))Screen quality - DAPI Count
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: Clear edge effects affecting total
cell count, however proportion of percentage positive ARG/INOS/BODIPY
seem unaffected by edge effect. Overall pattern of death is consistent
across plates.
# create a function to plot features
genheatmap <- function(feature){
# filter the dataframe
data_toplot <- data_norm_melt %>%
filter(Feature == feature) %>%
arrange(Condition_Name) %>%
mutate(Raw = case_when(grepl("Empty", Description) ~ NA, TRUE ~ Raw)) %>%
mutate(Row = as.numeric(factor(substr(WellID, 1, 1), levels = LETTERS[1:16])),
Column = as.numeric(substr(WellID, 2, 3)))
# create feature heading
cat("\n###", feature, "{.tabset .tabset-fade .tabset-pills} \n")
# generate a list to plot
plist <- list()
# create for loop to plot per condition (or cell line)
for (condition in unique(data_toplot$Condition_Name)){
data_plot <- data_toplot %>%
filter(Condition_Name == condition) #condition
# create for loop to plot per plate
for (i in unique(data_plot$Barcode)) {
plot_df <- data_plot %>%
filter(Barcode == i)
# Using geom tile to plot heat map
plist[[i]] <- plot_df %>%
ggplot(data = ., aes(x = Column, y = Row, fill = Raw)) +
geom_tile(aes(linewidth = ifelse(grepl("Media", Compound_Name), "Media", "Non-Media")),
colour = "black", width = 0.8, height = 0.8) +
ggtitle(paste0(unique(plot_df$Barcode), ": \n", unique(plot_df$PlateID))) +
scale_fill_gradient2(
low = "blue", mid = "white", high = "red", na.value = "grey80",
midpoint = median(plot_df$Raw, na.rm = TRUE), # decide on the max,med,min, here it is from plot_df
limits = c(min(plot_df$Raw, na.rm = TRUE), max(plot_df$Raw, na.rm = TRUE))) +
scale_linewidth_manual(values = c("Media" = 0.65, "Non-Media" = 0.3), guide = "none") +
scale_y_reverse(breaks = 1:16, labels = LETTERS[1:16]) + # Y-axis for rows
scale_x_continuous(breaks = 1:24, labels = sprintf("%02d", 1:24), position = "top") + # X-axis for columns
theme_bw() +
theme(
plot.title = element_text(size = 14),
axis.text.x = element_text(size = 7),
axis.text.y = element_text(size = 7),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
panel.grid = element_blank())
}
}
cat("\n")
#print all plots
wrap_plots(plist) + plot_layout(ncol = 2)
}
genheatmap("Total_DAPI_Count")Total_DAPI_Count
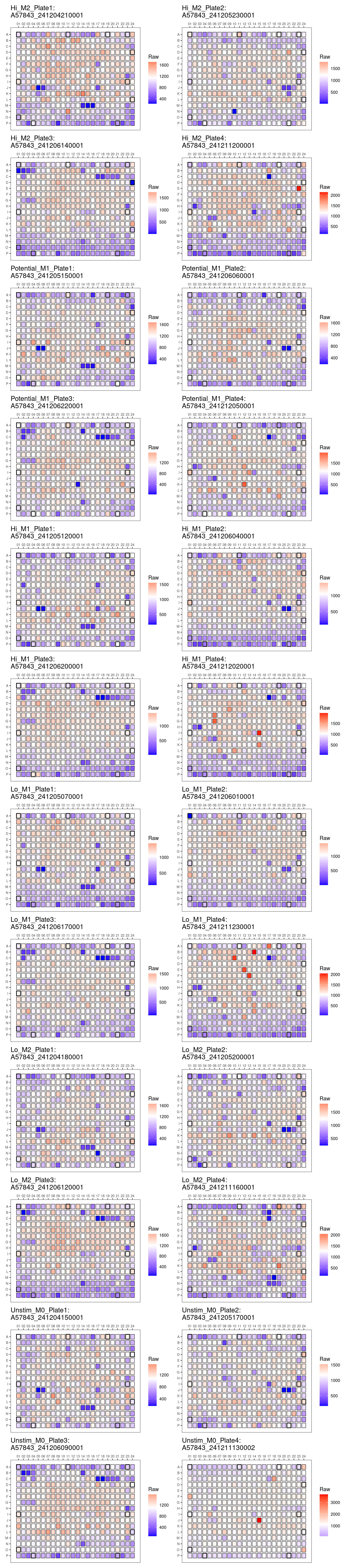
Plate QC Metrics
Comments:
DMSO: %CVs are
well within acceptable range except Hi_M2_Plate4 and Lo_M2_Plate4.
Unstim_DMSO: %CVs are well within acceptable range
except Hi_M2_Plate3 and Unstim_M0_Plate4.
Media:
%CVs are well within acceptable range except Hi_M2_Plate3.
Unstim_Media: %CVs are well within acceptable range
except Lo_M1_Plate2 and Lo_M2_Plate2.
Staurosporine: %CVs are high, particularly for 10uM
dose.
IL4+IL13: %CVs are within acceptable range
except Lo_M2_Plate4.
IL4+IL13_Unstim: %CVs are
within acceptable range except Hi_M2_Plate2, Lo_M2_Plate3.
LPS+INFg: %CVs are within acceptable range except
Lo_M2_Plate4.
LPS+INFg_Unstim: %CVs are within
acceptable range except Potential_M1_Plate2, Hi_M1_Plate1,
Lo_M1_Plate4.
2DG, Antimycin, Itaconate: %CVs are
high.
# filter down to control wells
ctrl_data <- ungroup(data_norm_melt) %>%
filter(Description %in% c("Negative_Control", "Positive_Control")) %>%
unite(Conc, c(Concentration, Units), sep = "") # Calculate summary statistics
screen_QC <- ctrl_data %>%
group_by(Condition_Name, Compound_Name, Conc, Feature) %>%
summarise_at(vars(Raw, NormToUnstimDMSO, NormToStimDMSO), lst(mean, sd), na.rm = TRUE) %>%
mutate(`NormToUnstimDMSO_%cv` = (`NormToUnstimDMSO_sd`/`NormToUnstimDMSO_mean`)*100)
write_csv(screen_QC, paste0("output/", prefix, "_screen_QC.csv"))
# Calculate summary statistics
perplate_QC <- ctrl_data %>%
group_by(Condition_Name, VCFG_Plate_ID, Compound_Name, Conc, Feature, Barcode) %>%
summarise_at(vars(Raw, NormToUnstimDMSO, NormToStimDMSO), lst(mean, sd), na.rm = TRUE) %>%
mutate(`NormToUnstimDMSO_%cv` = (`NormToUnstimDMSO_sd`/`NormToUnstimDMSO_mean`)*100)
write_csv(perplate_QC, paste0("output/", prefix, "_perplate_QC.csv"))# create function to generate datatable
condition_list <- unique(perplate_QC$Condition_Name)
genplateqc <- function(feature){
# print feature heading
cat("\n### ", feature, "{.tabset .tabset-fade .tabset-pills}\n")
# for loop to create condition plates
for(condition in condition_list){
cat("\n#### ", condition, "\n")
perplate_QC_filt <- ungroup(perplate_QC) %>%
filter(Feature == feature & Condition_Name == condition) %>%
unite(Control, c(Compound_Name, Conc)) %>%
mutate_if(is.numeric, round, 2) %>%
select(-Condition_Name, -Feature)
# display the data in a table
print(htmltools::tagList(datatable(
perplate_QC_filt, extensions = c('Buttons', 'Scroller', "FixedColumns", 'ColReorder'), rownames = FALSE,
options = list(
dom = 'Blrtip',
columnDefs = list(list(className = 'dt-center',targets="_all")),
lengthMenu = list(c(5, -1), c("5", "All")),
pageLength = 5,
buttons = list(list(extend = 'csv',
filename = paste0(prefix, "_PerPlateQC_", feature, "_", condition)),
list(extend = 'excel',
filename = paste0(prefix, "_PerPlateQC_", feature, "_", condition), title = NULL)),
searching = TRUE,
scrollX = TRUE,
fixedColumns = list(leftColumns = 2))) %>%
formatStyle(c("NormToUnstimDMSO_%cv"), Color = styleInterval(24, c("Black", "red")),
fontWeight = styleInterval(24, c("bold", "bold")))))
cat("\n")
}
cat("\n")
}
genplateqc("Total_DAPI_Count")Total_DAPI_Count
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small number
of outliers, but values are very tight overall.
#create a list of features
feat_list <- unique(ctrl_data$Feature)
ctrl_data_plot <- ctrl_data %>%
unite(Control, c(Compound_Name, Conc), remove = FALSE)
# set order of compounds for plotting
ctrl_order <- c("DMSO_Unstim_0.1%", "DMSO_0.1%", "Media_Unstim_0uM", "Media_0uM",
"INFg_Unstim_25ng/mL", "INFg_25ng/mL",
"IL4+IL13_50ng/mL", "IL4+IL13_Unstim_50ng/mL",
"LPS+INFg_50ng/mL", "LPS+INFg_Unstim_50ng/mL",
"2DG_0.5mM", "2DG_1mM", "2DG_2mM",
"Antimycin_0.63uM", "Antimycin_1.25uM", "Antimycin_2.5uM",
"Itaconate_0.5mM", "Itaconate_1mM", "Itaconate_2mM",
"Staurosporine_0.1uM", "Staurosporine_1uM", "Staurosporine_10uM")
cond_labs_order <- c("Unstimulated", "25 ng/mL INFg",
"3 ng/mL INFg + 3 ng/mL LPS", "25 ng/mL INFg + 25 ng/mL LPS",
"5 ng/mL IL4 + 5 ng/mL IL13", "10 ng/mL IL4 + 10 ng/mL IL13")
ctrl_data_plot$Control <- factor(ctrl_data_plot$Control, levels = ctrl_order)
ctrl_data_plot$Condition_Name <- factor(ctrl_data_plot$Condition_Name, levels = cond_labs_order)
# create box plots with outliers
genboxplot <- function(compounds, vars){
ctrl_data_plot %>%
filter(grepl(compounds, Compound_Name) & Feature == vars) %>%
ggplot(., aes(x = Control,
y = Raw,
fill = Control)) +
geom_boxplot(alpha = 0.7, notch = FALSE, outlier.colour = "black") +
facet_grid(Condition_Name+Phenotype~factor(Stimulation, levels = c("Unstimulated", "Stimulated"))) +
labs(title = paste0("\n", vars, "\n"),
y = paste0("\n", vars)) +
theme_bw() +
theme(plot.title = element_text(size = 13),
axis.title.x = element_blank(),
axis.title.y = element_text(size = 11, margin=margin(0,10,0,0)),
axis.text.x = element_text(hjust = 1, vjust = 1.1, size = 11, angle = 45, margin=margin(10,0,0,0), colour = "black"),
axis.text.y = element_text(size = 10, margin=margin(0,10,10,0), colour = "black"),
strip.text = element_text(size = 12),
legend.position = "none")
}
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[1])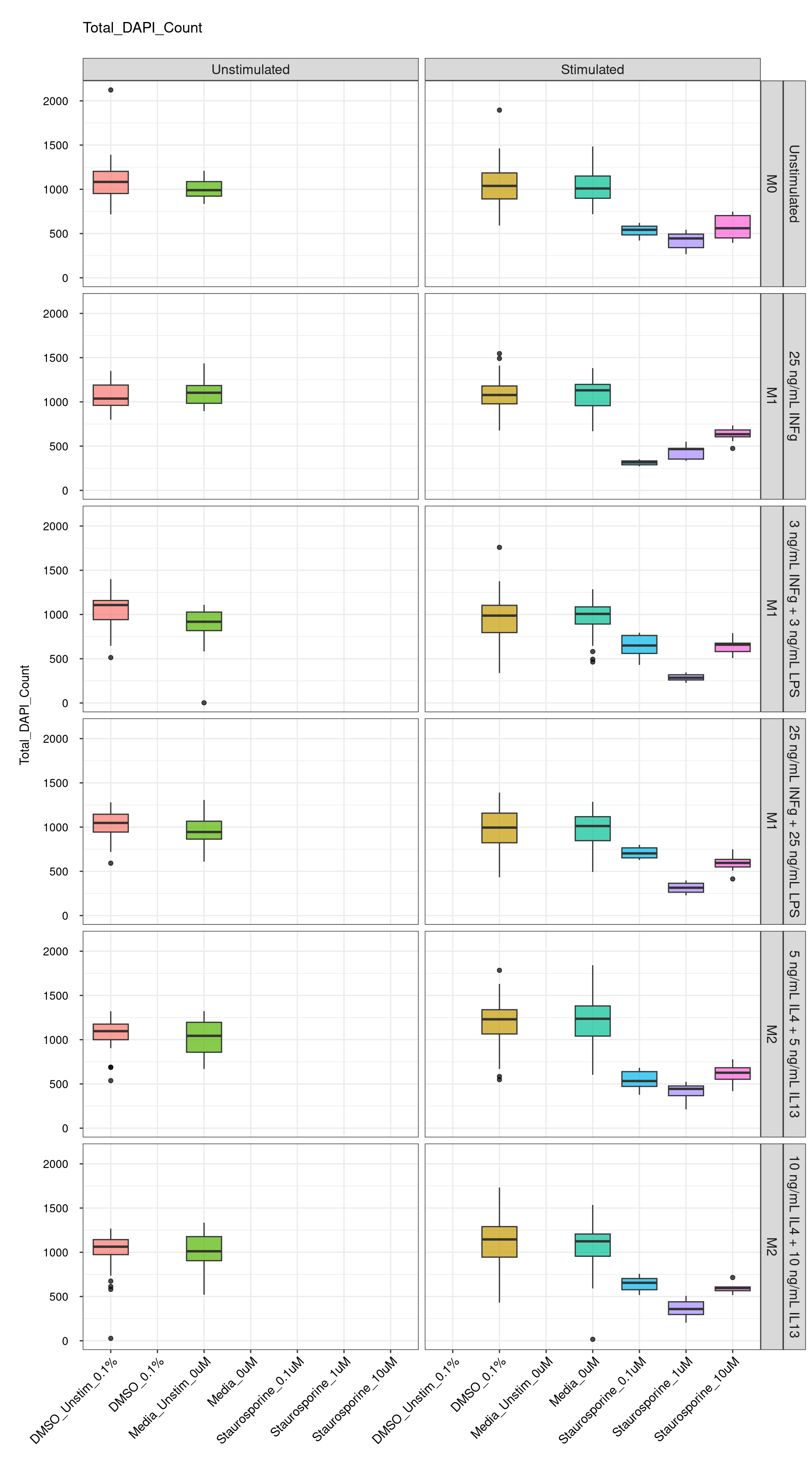
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[1])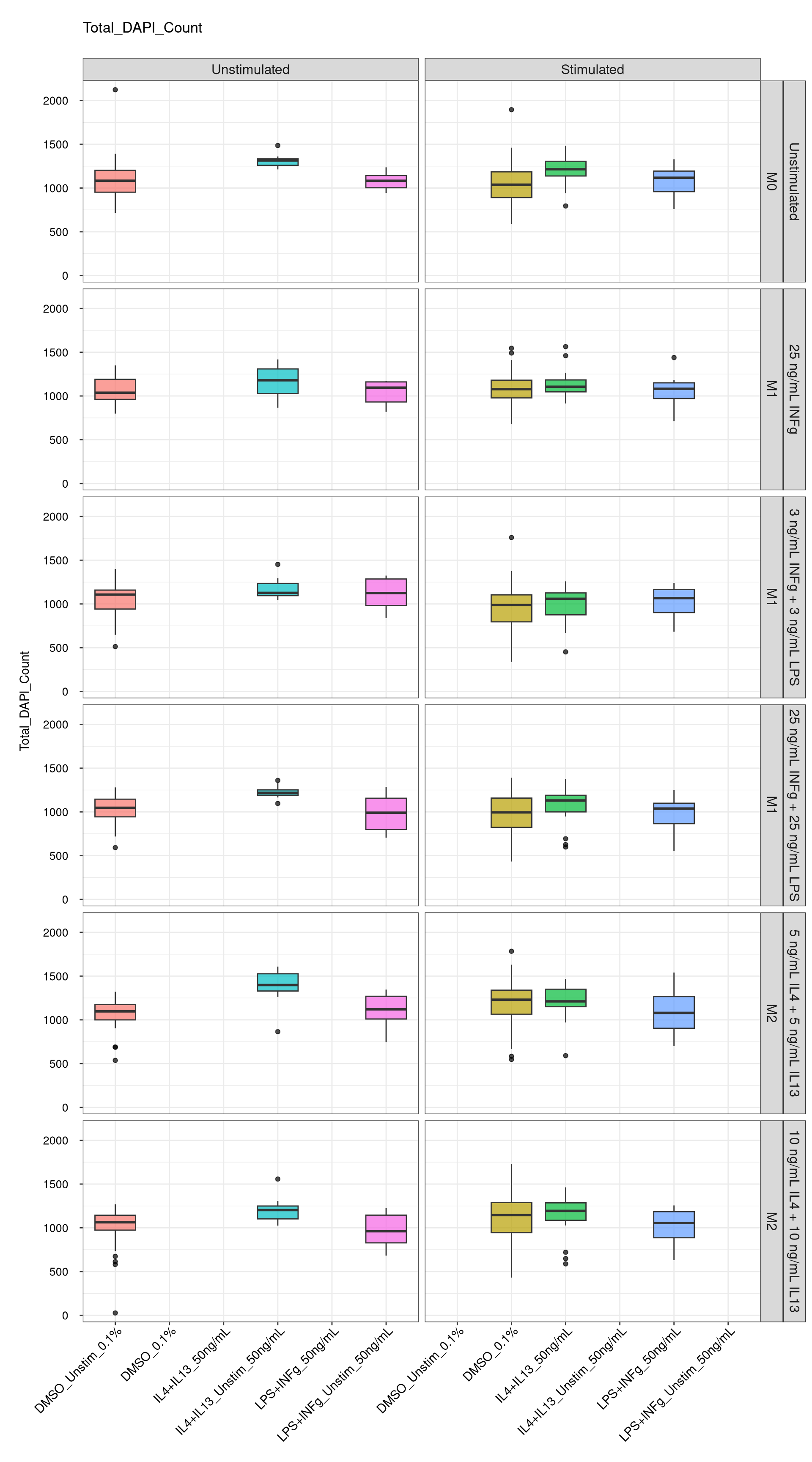
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[1])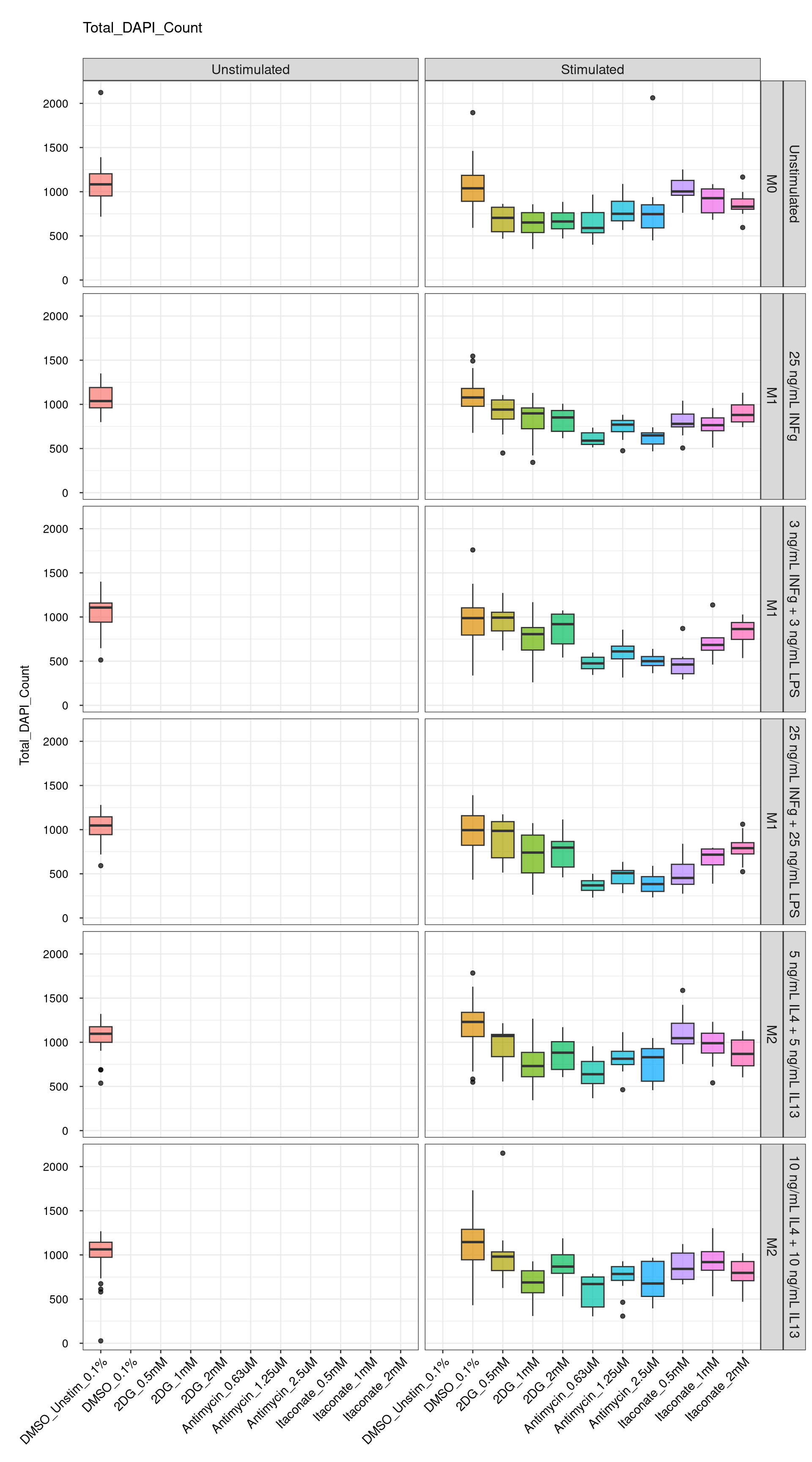
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# prepare the data for plotting
# unite columns to create unique ID
normplotting <- ungroup(data_norm_melt) %>%
# filter(Cell_Line == Cell_Line) %>%
unite("Plate", c("Barcode", "WellID"), sep = "_", remove = FALSE) %>%
unite(Conc, c(Concentration, Units), remove = FALSE, sep = "") %>%
unite(Control, c(Compound_Name, Conc), remove = FALSE) %>%
mutate(Category = case_when(Description == "Sample" ~ "Sample",
Description == "Excluded" ~ "Excluded",
TRUE ~ Control)) %>%
mutate_if(is.numeric, round, 2) %>%
arrange(Barcode, WellID)
# set order of compounds for plotting
cat_order <- c("Sample", "Excluded", "DMSO_Unstim_0.1%", "DMSO_0.1%", "Media_Unstim_0uM", "Media_0uM",
"INFg_Unstim_25ng/mL", "INFg_25ng/mL",
"IL4+IL13_50ng/mL", "IL4+IL13_Unstim_50ng/mL", "LPS+INFg_50ng/mL", "LPS+INFg_Unstim_50ng/mL",
# "IL4+IL13_Unstim_0ng/mL", "IL4+IL13_0ng/mL", "IL4+IL13_Unstim_5ng/mL", "IL4+IL13_5ng/mL", "IL4+IL13_Unstim_10ng/mL", "IL4+IL13_10ng/mL",
# "LPS+INFg_Unstim_0ng/mL", "LPS+INFg_0ng/mL", "LPS+INFg_Unstim_3ng/mL", "LPS+INFg_3ng/mL", "LPS+INFg_Unstim_25ng/mL", "LPS+INFg_25ng/mL",
"2DG_0.5mM", "2DG_1mM", "2DG_2mM",
"Antimycin_0.63uM", "Antimycin_1.25uM", "Antimycin_2.5uM",
"Itaconate_0.5mM", "Itaconate_1mM", "Itaconate_2mM",
"Staurosporine_0.1uM", "Staurosporine_1uM", "Staurosporine_10uM")
cond_labs_order <- c("Unstimulated", "25 ng/mL INFg",
"3 ng/mL INFg + 3 ng/mL LPS", "25 ng/mL INFg + 25 ng/mL LPS",
"5 ng/mL IL4 + 5 ng/mL IL13", "10 ng/mL IL4 + 10 ng/mL IL13")
normplotting$Category <- factor(normplotting$Category, levels = cat_order)
# emulate the ggplot colour palette so test compounds can be manually coloured grey
# create the function
gg_color <- function(n) {
hues = seq(15, 375, length = n + 1)
hcl(h = hues, l = 65, c = 100)[1:n]
}
# call the first n colours
n = length(unique(normplotting$Category)) + 1
cols = gg_color(n)
y <- 1:length(unique(normplotting$Category))
# Define the feature
feature_list <- unique(normplotting$Feature)
value_list <- colnames(normplotting[c(19, 20, 21)])
cond_list <- cond_labs_order
# plot dot plot function
gen_dotplot <- function(feature, values, condition) {
normplotting %>%
filter(Feature == feature & Condition_Name == condition) %>%
ggplot(., aes(x = WellID,
y = .data[[values]],
label = Compound_Name)) +
geom_point(aes(colour = Category,
text = paste('Compound: ', Compound_Name,'<br> WellID:', WellID, '<br> Value:', .data[[values]])),
alpha = 0.8, size = 1.6) +
facet_wrap(. ~ Barcode, nrow = 1) +
# facet_wrap(. ~ Hours, labeller = labeller(Hours = ~ paste(.x, "hours"))) +
labs(y = paste0(values)) +
theme_bw() +
theme(axis.title.y = element_text(margin = margin(t = 0, r = 0, b = 10, l = 10)),
axis.title.x = element_blank(),
axis.text.x = element_text(hjust = 1, vjust = 1.1, size = 10, angle = 45, margin=margin(10,0,0,0), colour = "black"),
axis.text.y = element_text(colour = "black"),
plot.margin = margin(t = 0, r = 10, b = 10, l = 10),
legend.title = element_blank(),
legend.background = element_blank(),
legend.box.background = element_rect(fill = "transparent", colour = "transparent"),
legend.key = element_rect(fill = "transparent", colour = "transparent"),
legend.text = element_text(size = 7)) +
scale_x_discrete(labels = "") +
scale_colour_manual(name = "", values = c("grey80", "grey50", cols), drop = FALSE)
}
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[1], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[1], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[1], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Screen quality - ARG Positive Perc
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: No obvious edge effects. Overall
pattern of death is consistent across plates. Consistently greater
dynamic range of raw and positive ARG percentages in M2 plates compared
to M1.
genheatmap("Positive_Perc_ARG")Positive_Perc_ARG
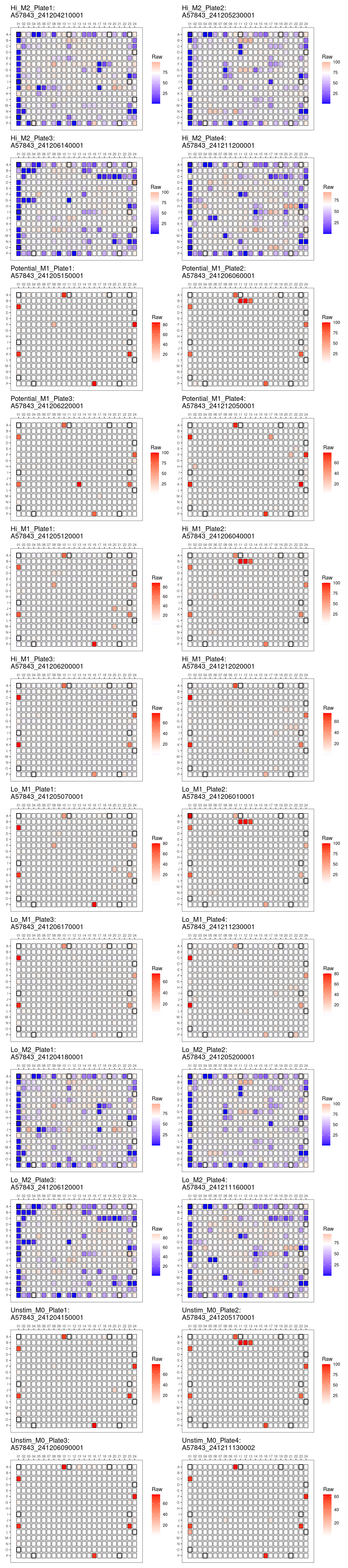
Plate QC Metrics
Comments:
Stimulated DMSO &
Stimulated Media: %CVs are well within acceptable limits on all
M2 plates.
Stimulated IL4+IL13: %CVs are well
within acceptable limits except on Hi_M1_Plate4, Lo_M1_Plate1,
Lo_M1_Plate3.
Unstimulated DMSO, Unstimulated Media,
LPS+INFg, Staurosporine: %CVs exceed acceptable limits but this
is to be expected given the low ARG positive percentages.
genplateqc("Positive_Perc_ARG")Positive_Perc_ARG
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small
number of outliers, but values are very tight overall.
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[2])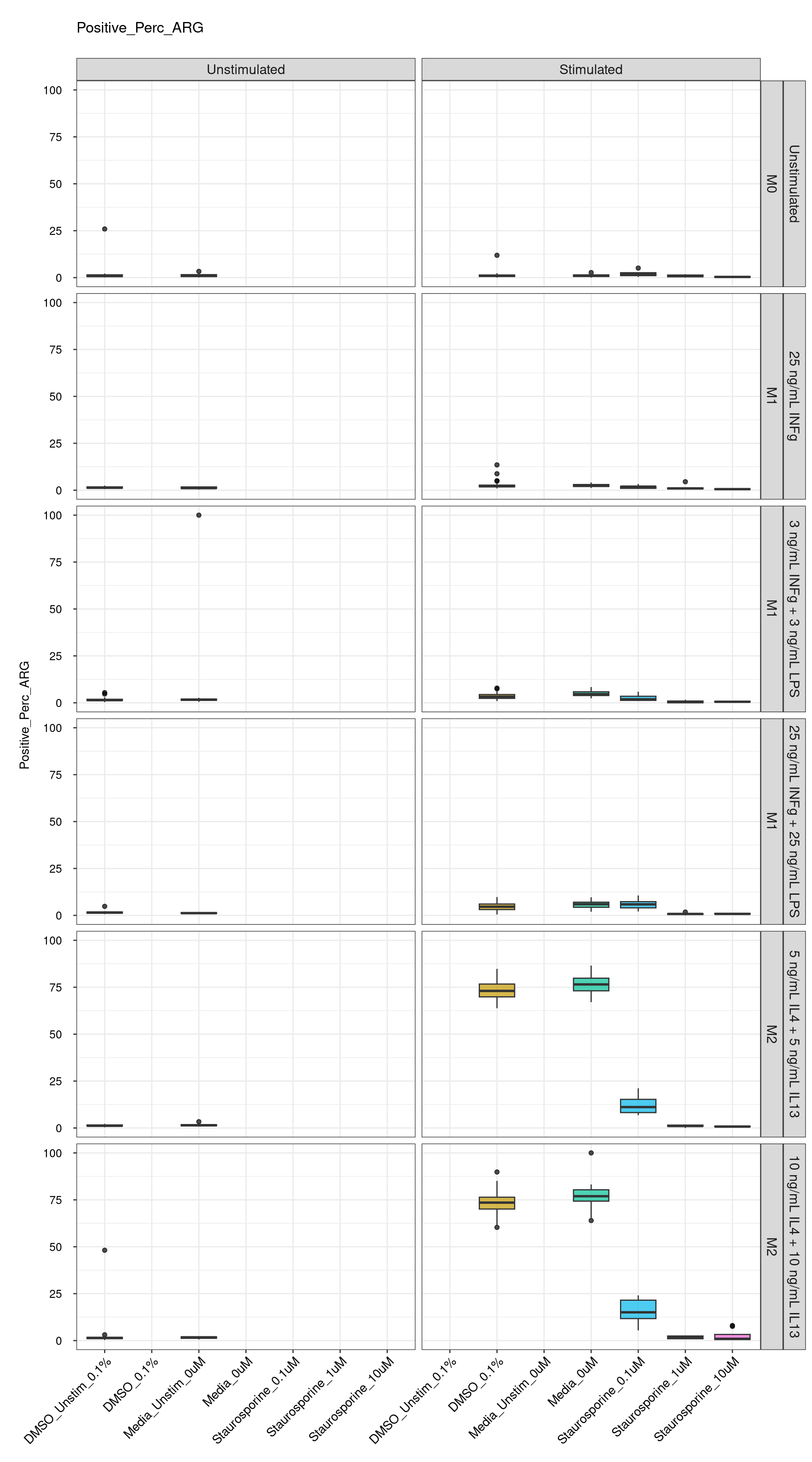
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[2])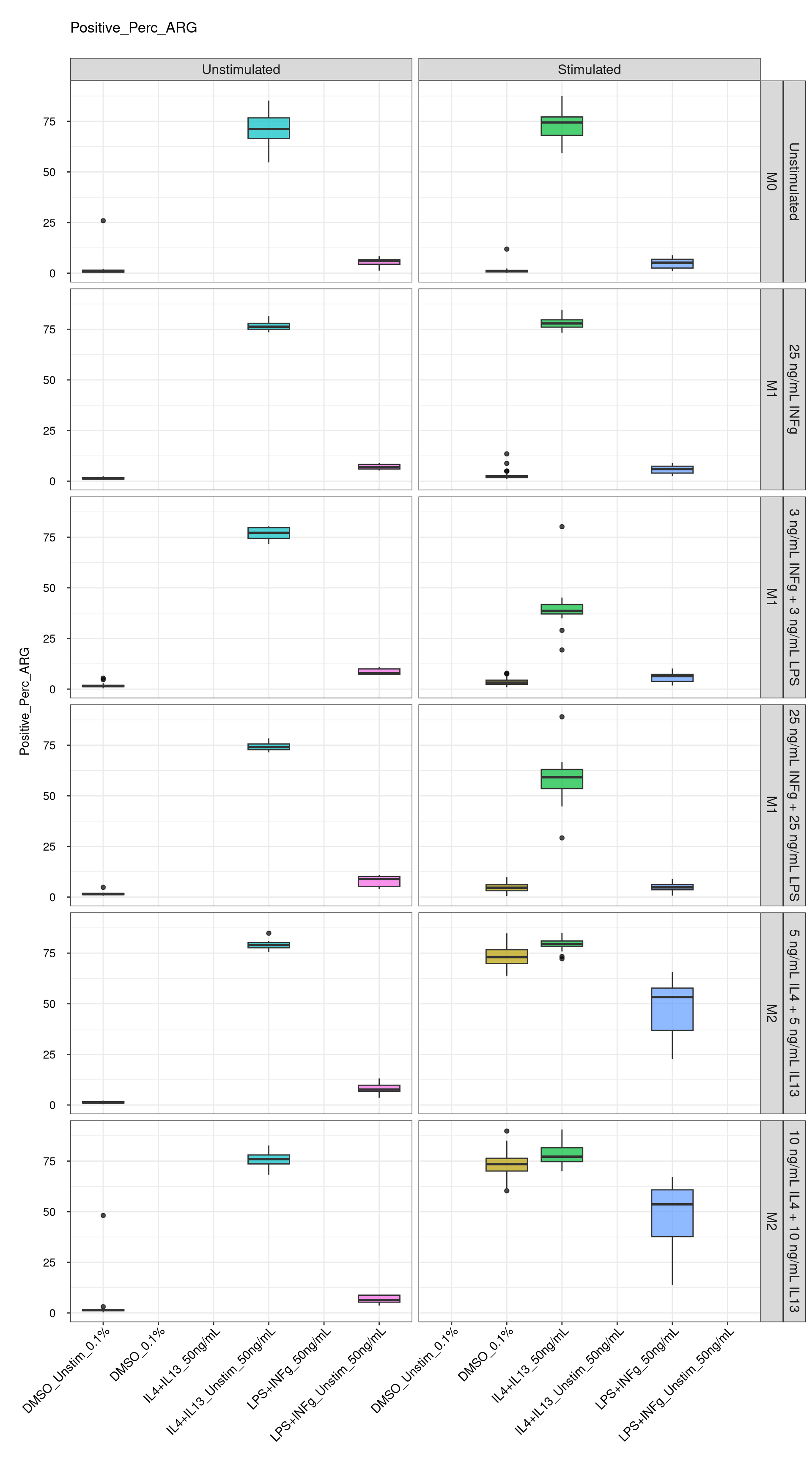
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[2])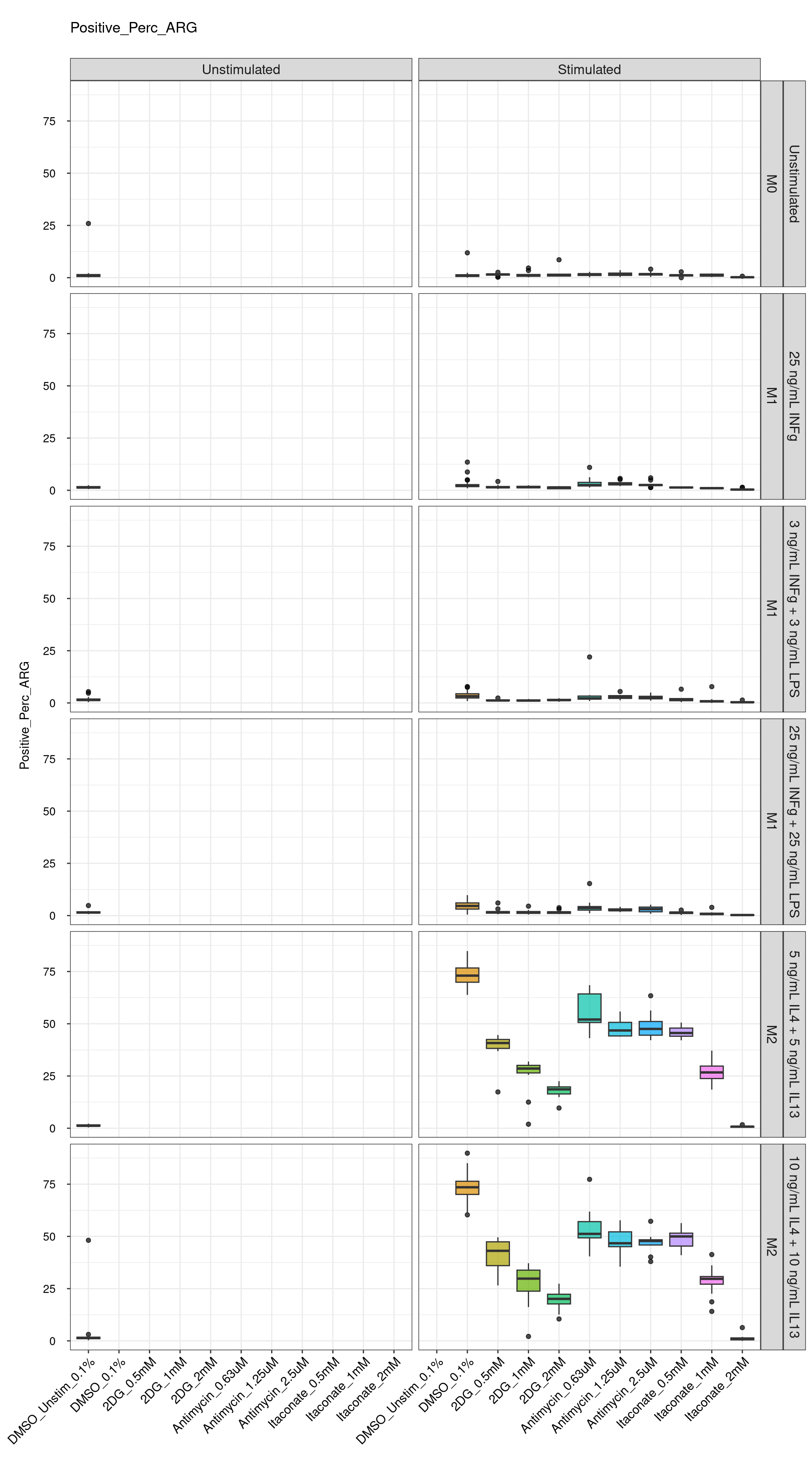
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[2], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[2], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[2], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Screen quality - INOS Positive Perc
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: No obvious edge effects. Overall
pattern of death is consistent across plates except
Unstimulated_M0_Plate4. Consistently greater dynamic range of raw and
positive INOS percentages in M1 plates compared to M2.
genheatmap("Positive_Perc_INOS")Positive_Perc_INOS

Plate QC Metrics
Comments:
Stimulated DMSO &
Stimulated Media: %CVs are well within acceptable limits on all
Lo and Hi M1 plates.
Stimulated LPS+INFg: %CVs are
well within acceptable limits except on all plates.
Unstimulated DMSO, Unstimulated Media, IL4+IL13,
Staurosporine: %CVs exceed acceptable limits but this is to be
expected given the low INOS positive percentages.
genplateqc("Positive_Perc_INOS")Positive_Perc_INOS
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small number
of outliers, but values are very tight overall.
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[3])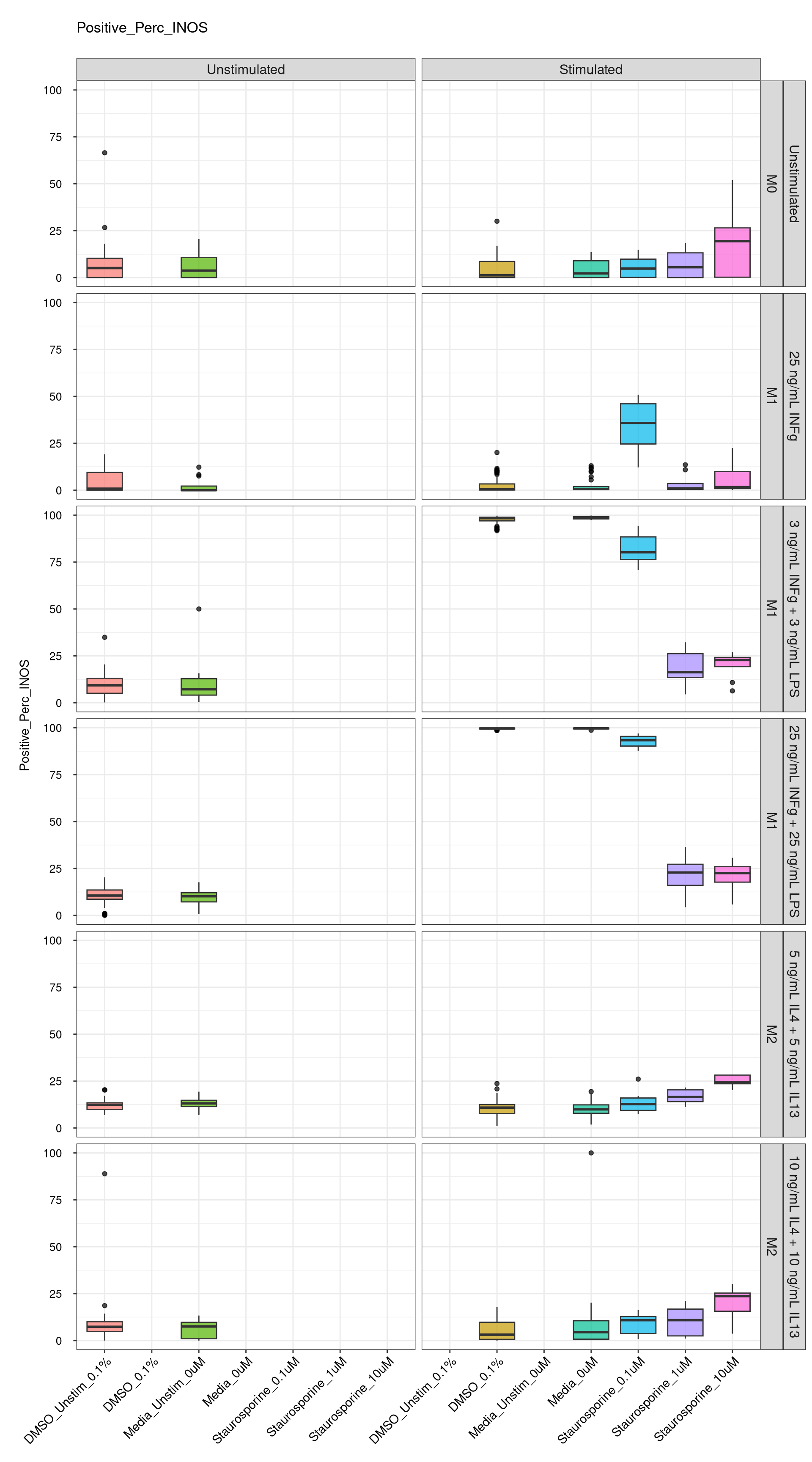
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[3])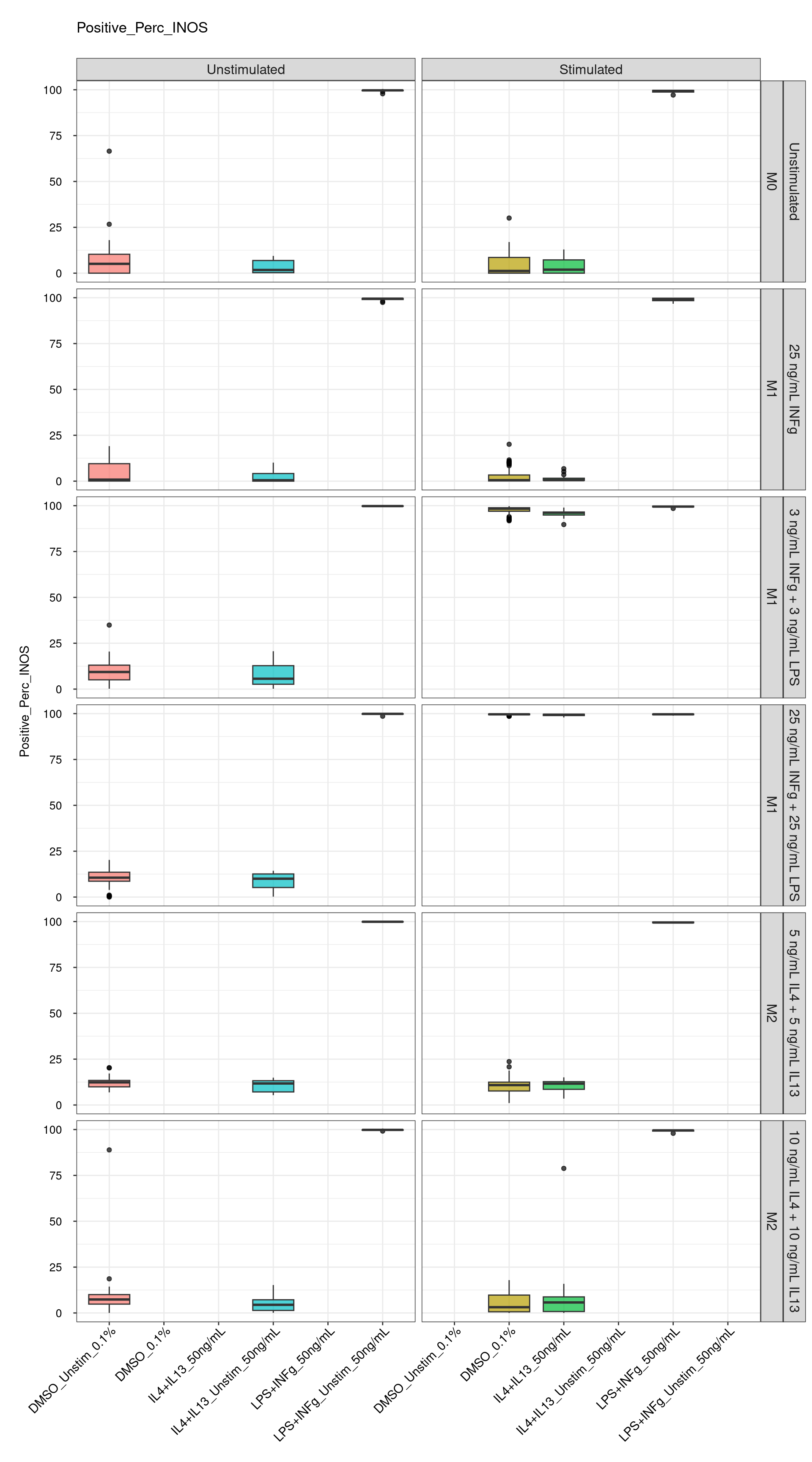
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[3])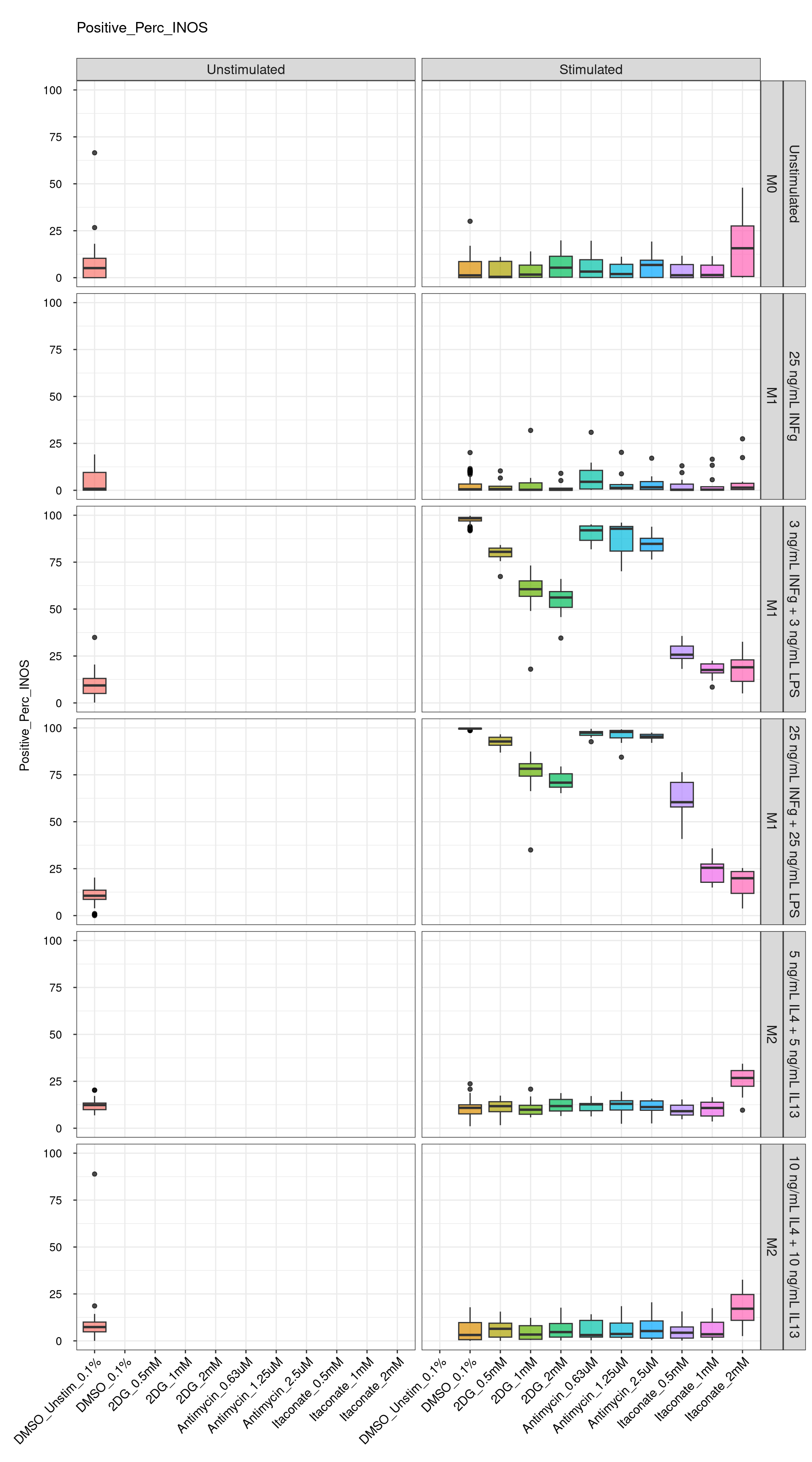
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[3], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[3], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[3], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Screen quality - INOS + BODIPY Positive Perc
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: No obvious edge effects. Overall
pattern of death is consistent across plates. Consistently greater
dynamic range of raw and positive INOS+BODIPY percentages in M1 plates
compared to M2.
genheatmap("Positive_Perc_INOS+BODIPY")Positive_Perc_INOS+BODIPY

Plate QC Metrics
Comments:
Stimulated DMSO &
Stimulated Media: %CVs are well within acceptable limits on all
Lo and Hi M1 plates except Lo_M1_Plate4 for Stimulated DMSO which is
just above what would be considered acceptable.
Stimulated
LPS+INFg: %CVs are well within acceptable limits except on all
plates.
Unstimulated DMSO, Unstimulated Media, IL4+IL13,
Staurosporine: %CVs exceed acceptable limits but this is to be
expected given the low INOS+BODIPY positive percentages.
genplateqc("Positive_Perc_INOS+BODIPY")Positive_Perc_INOS+BODIPY
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small number
of outliers, but values are very tight overall.
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[4])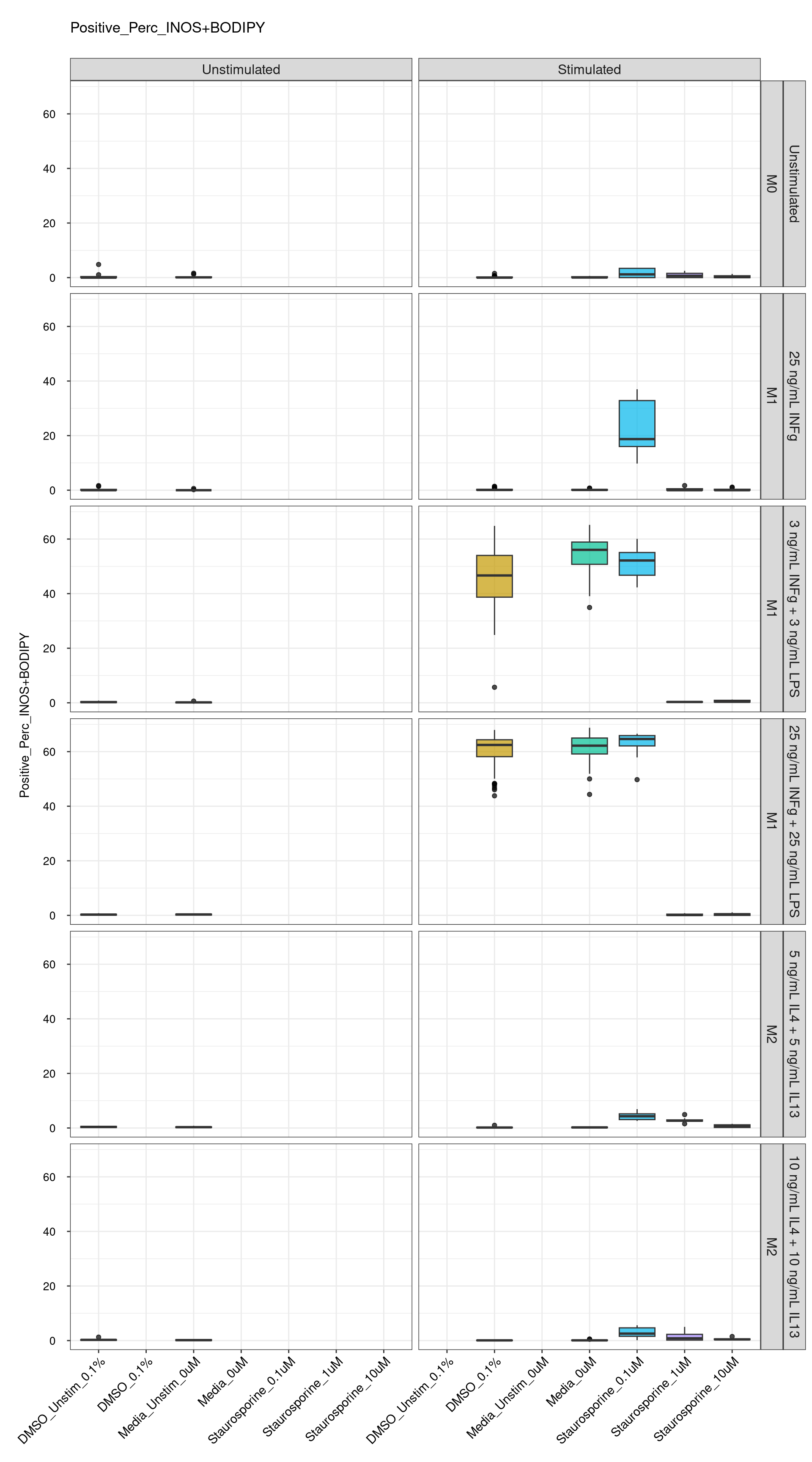
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[4])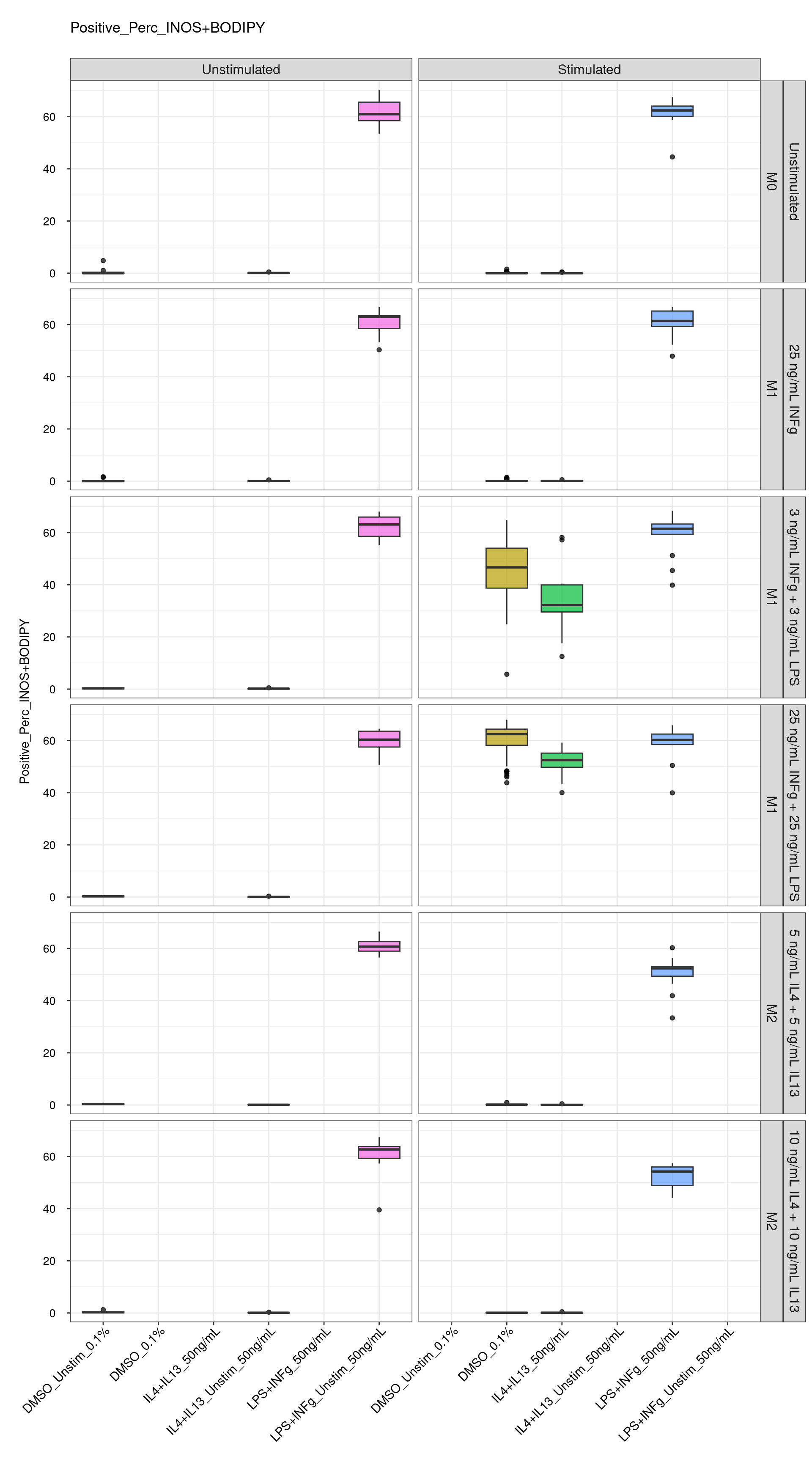
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[4])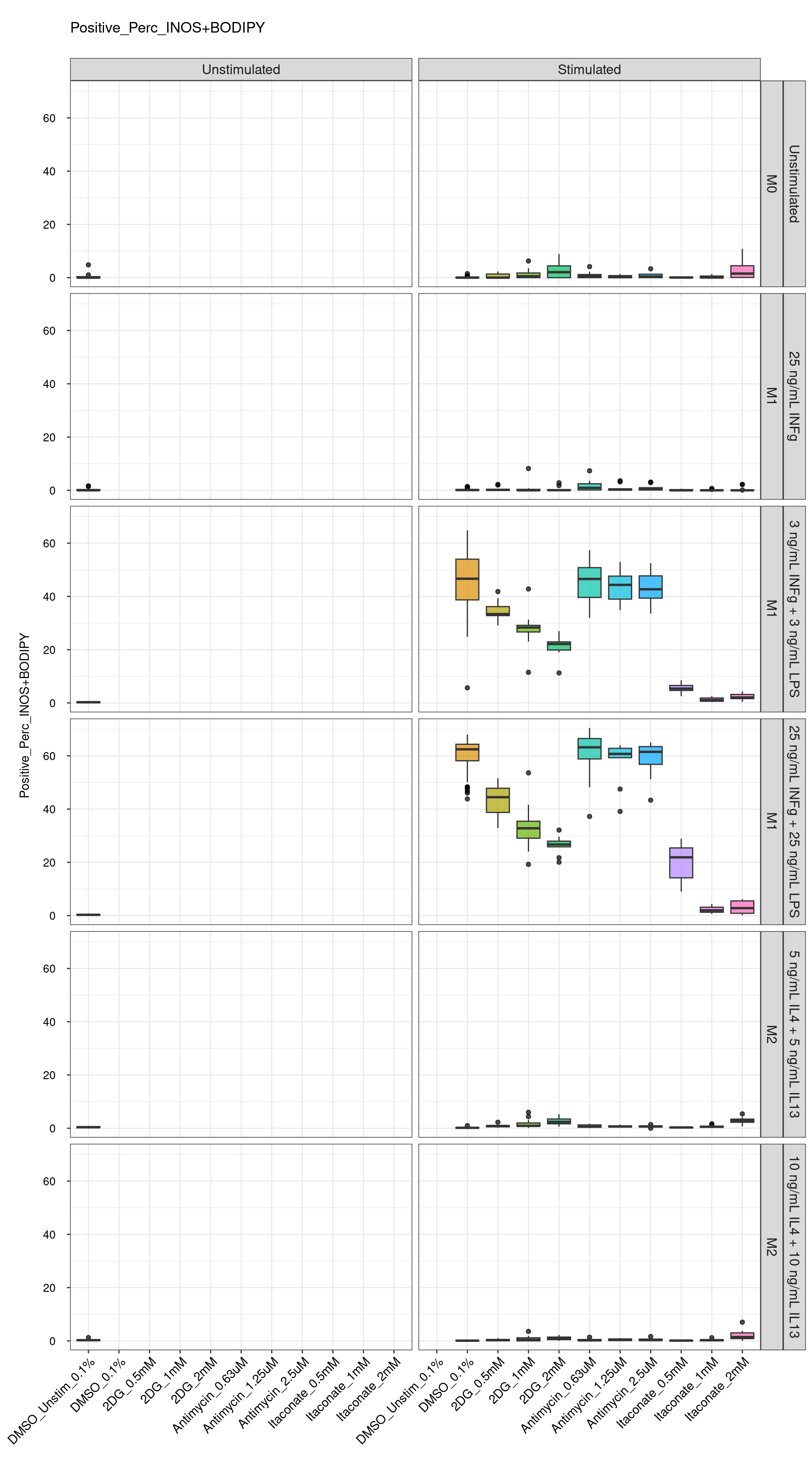
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[4], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[4], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[4], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Screen quality - INOS - BODIPY Positive Perc
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: No obvious edge effects. Overall
pattern of death is consistent across plates. Consistently greater
dynamic range of raw and positive INOS-BODIPY percentages in M1 plates
compared to M2.
genheatmap("Positive_Perc_INOS-BODIPY")Positive_Perc_INOS-BODIPY
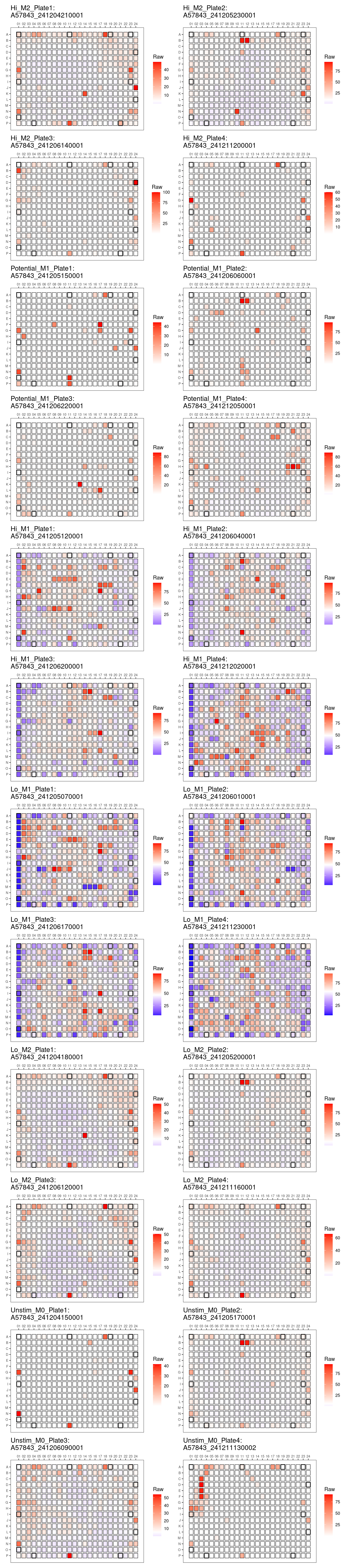
Plate QC Metrics
Comments:
Stimulated DMSO &
Stimulated Media: %CVs are well within acceptable limits on all
Lo and Hi M1 plates.
Stimulated LPS+INFg: %CVs are
well within acceptable limits on all plates.
Unstimulated
DMSO, Unstimulated Media, IL4+IL13, Staurosporine: %CVs exceed
acceptable limits but this is to be expected given the low INOS-BODIPY
positive percentages.
genplateqc("Positive_Perc_INOS-BODIPY")Positive_Perc_INOS-BODIPY
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small number
of outliers, but values are very tight overall.
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[5])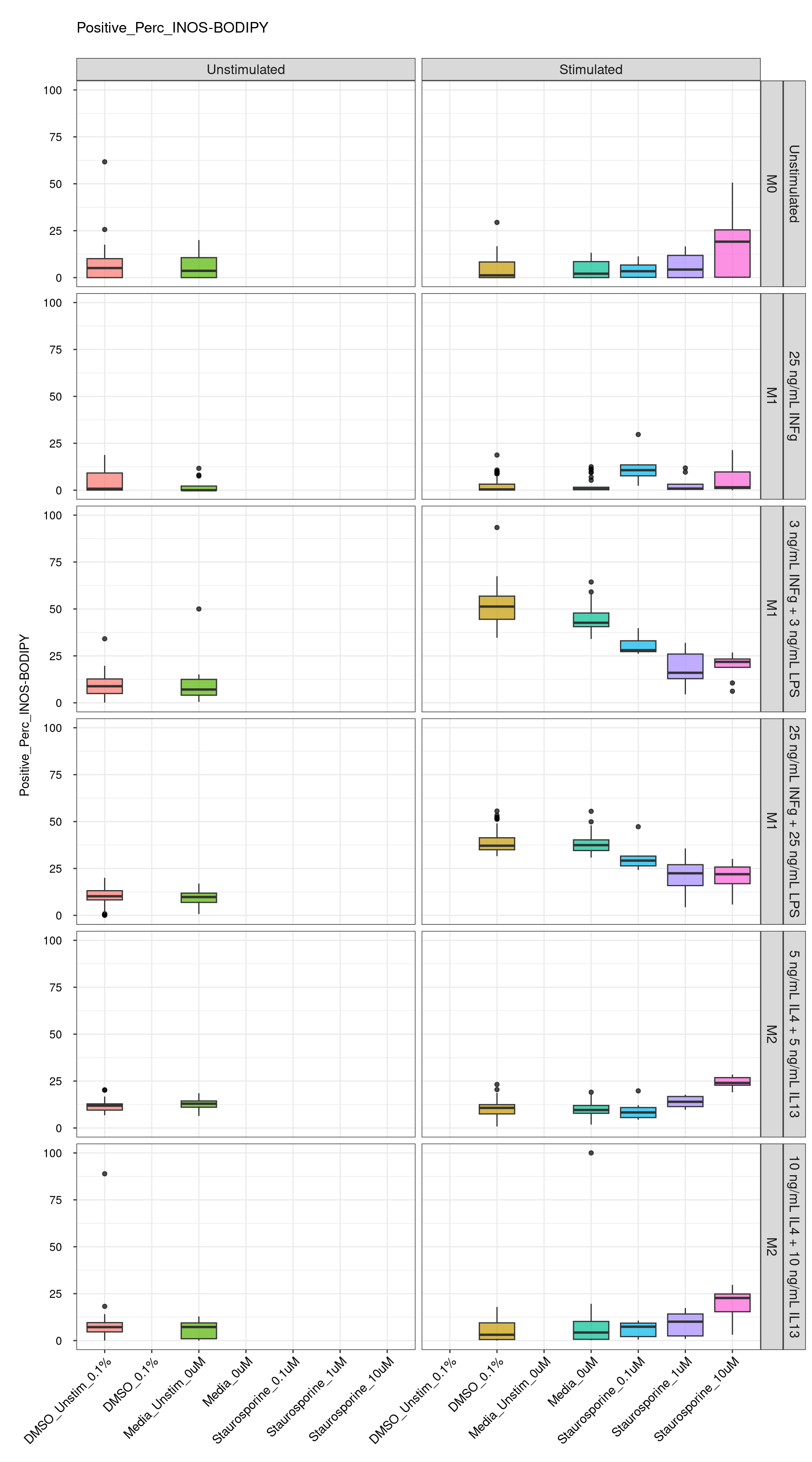
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[5])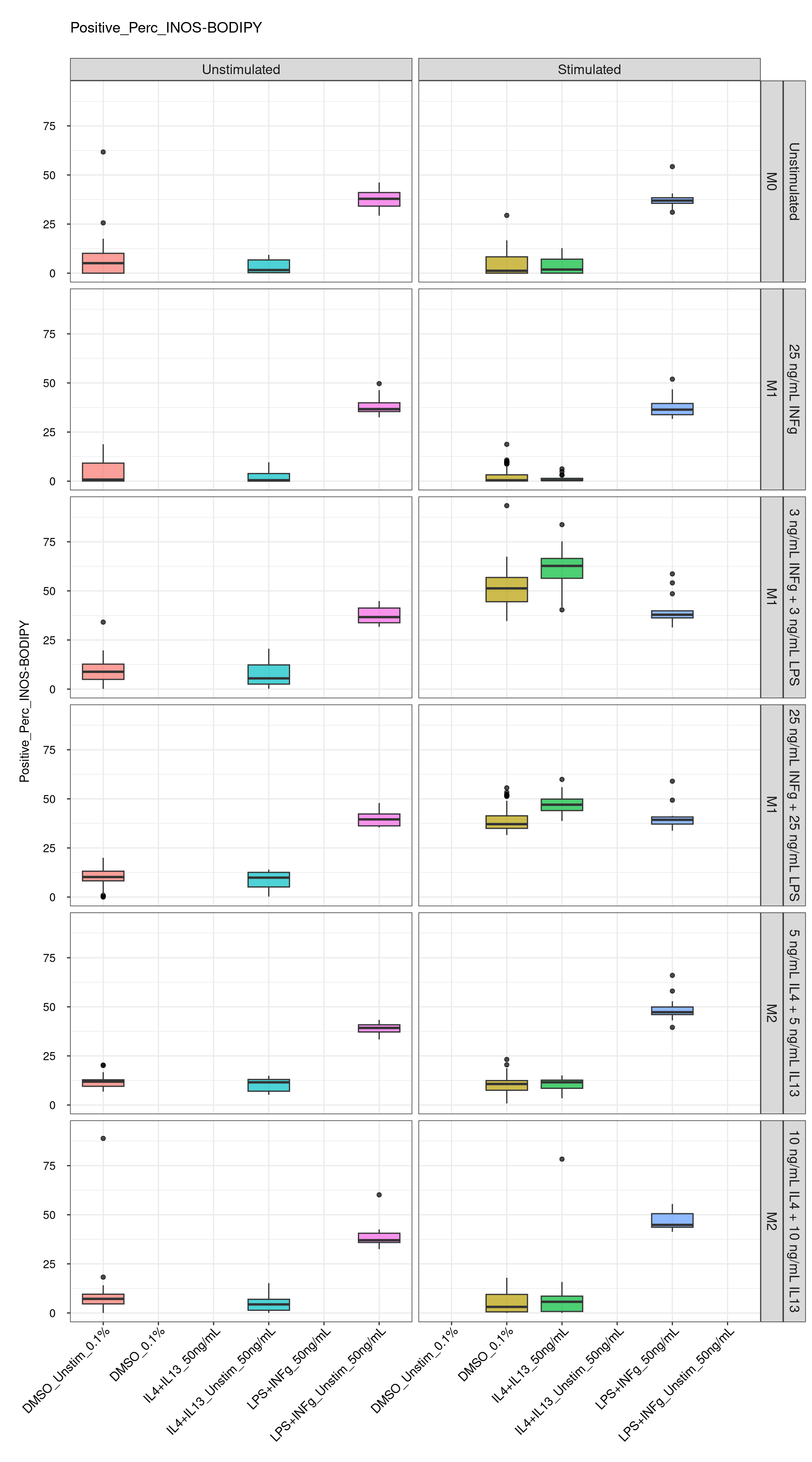
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[5])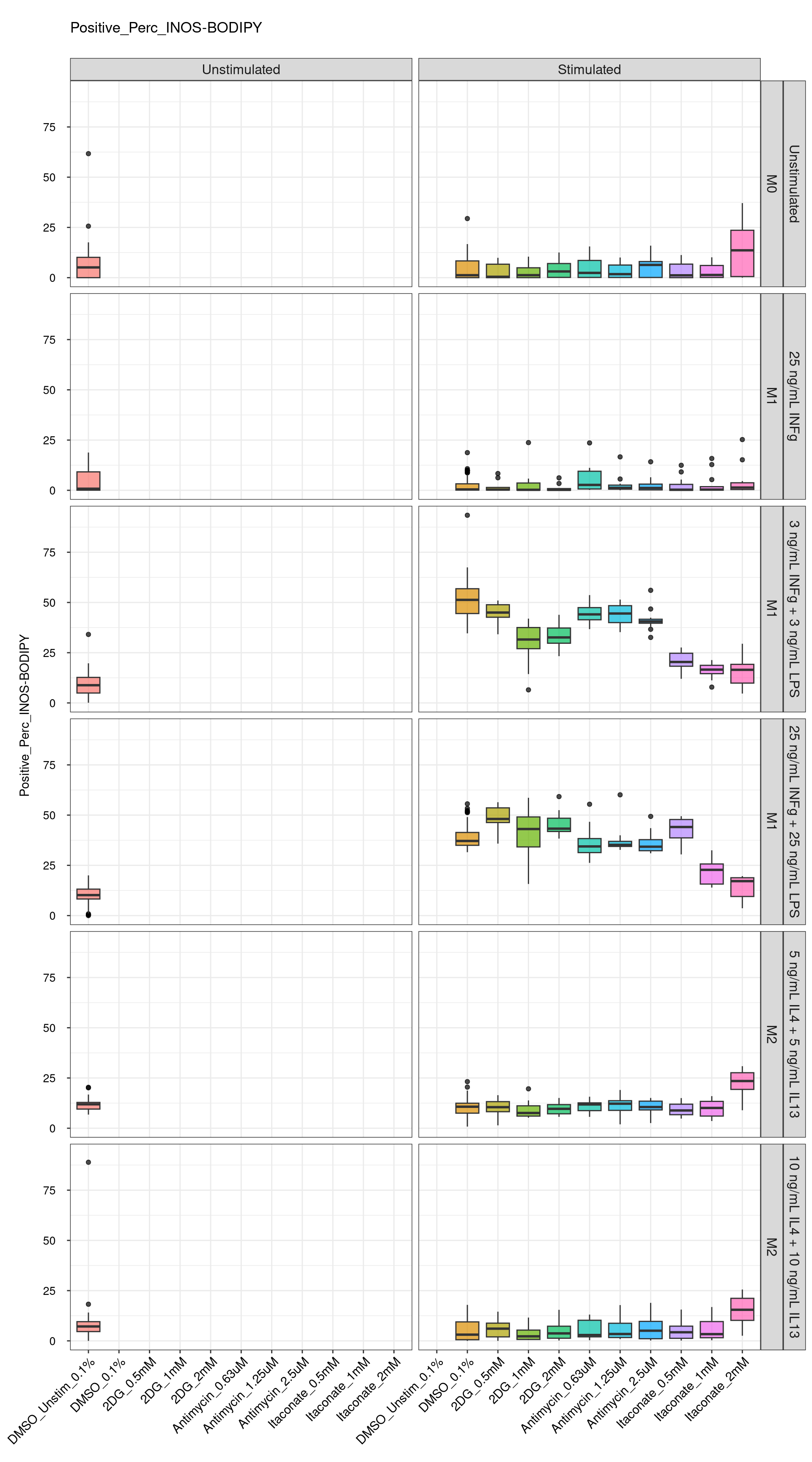
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[5], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[5], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[5], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Screen quality - Mean Spot Count Per Cell (BODIPY)
See the Screen quality section of the
Methods page for a more information regarding what’s expected in terms
of heat maps and screen and plate QC metrics, including %CVs and Z’
Factor values.
Heat maps
Comments: Clear edge effects affecting total
cell count, however proportion of percentage positive ARG/INOS/BODIPY
seem unaffected by edge effect. Overall pattern of death is consistent
across plates.
genheatmap("MeanSpotCount_PerCell")MeanSpotCount_PerCell

Plate QC Metrics
Comments:
Stimulated DMSO &
Stimulated Media: %CVs are well within acceptable limits.
Stimulated LPS+INFg: %CVs are well within acceptable
limits
Unstimulated DMSO, Unstimulated Media, IL4+IL13,
Staurosporine: %CVs exceed acceptable limits but this is to be
expected given the low BODIPY counts.
genplateqc("MeanSpotCount_PerCell")MeanSpotCount_PerCell
10 ng/mL IL4 + 10 ng/mL IL13
25 ng/mL INFg
25 ng/mL INFg + 25 ng/mL LPS
3 ng/mL INFg + 3 ng/mL LPS
5 ng/mL IL4 + 5 ng/mL IL13
Unstimulated
Box plots
Comments:
DMSO - Small number
of outliers, but values are very tight overall.
cat("\n<br>\n")cat("\n### Negative Control \n")Negative Control
genboxplot("DMSO|Media|Staurosporine", feat_list[7])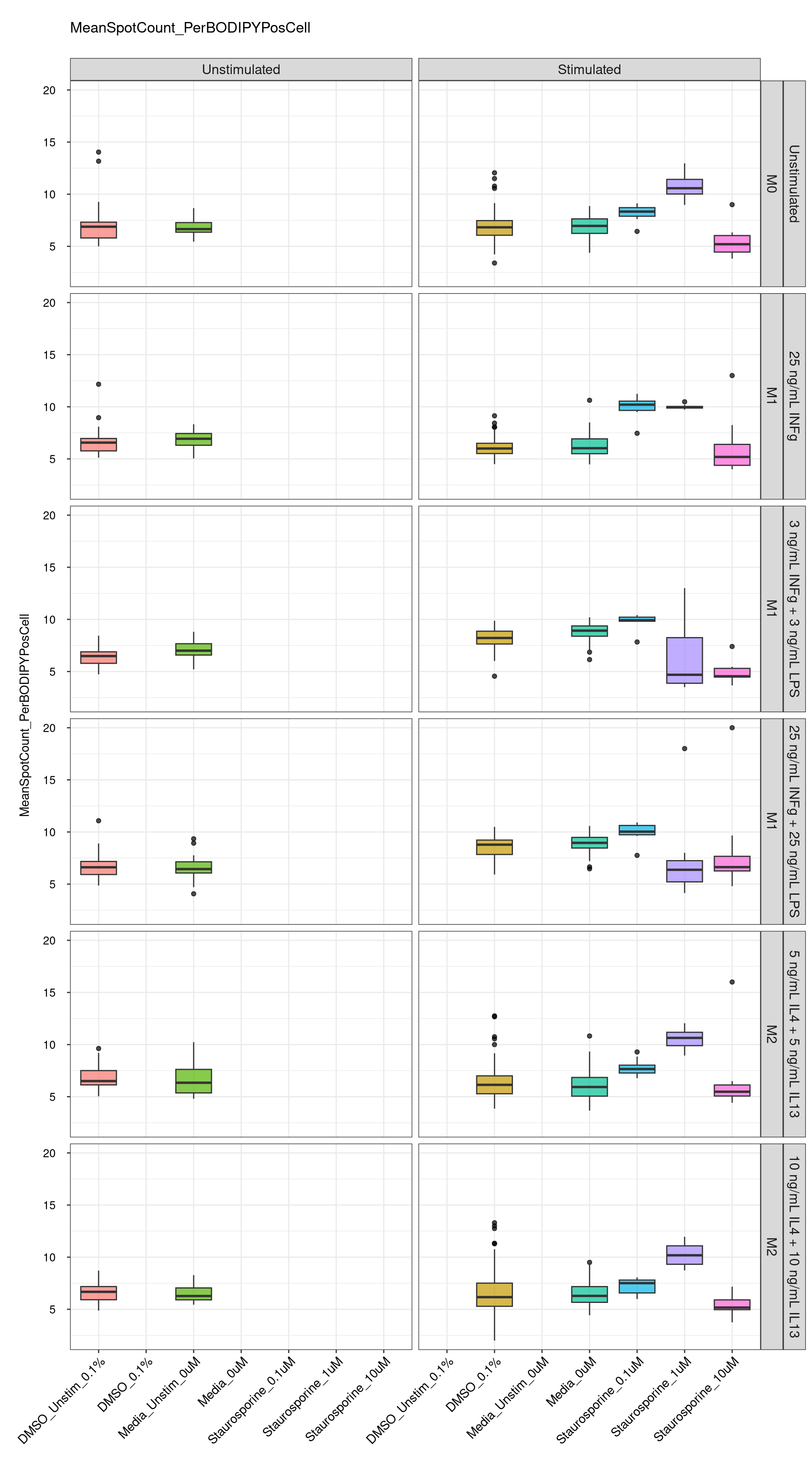
cat("\n<br>\n")cat("\n### Positive Control \n")Positive Control
genboxplot("DMSO|LPS|IL4", feat_list[7])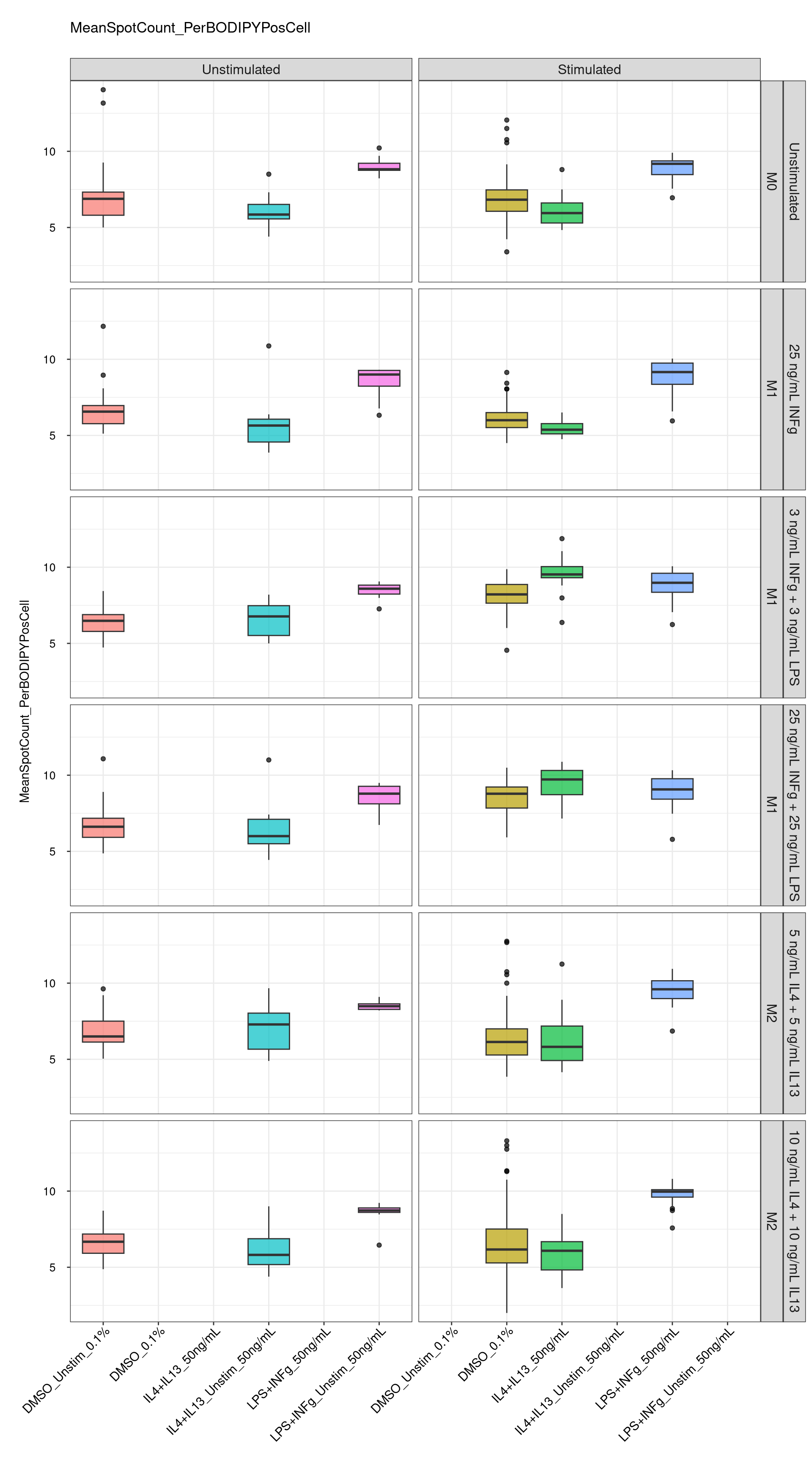
cat("\n<br>\n")cat("\n### Technical Control \n")Technical Control
genboxplot("DMSO|2DG|Antimycin|Itaconate", feat_list[7])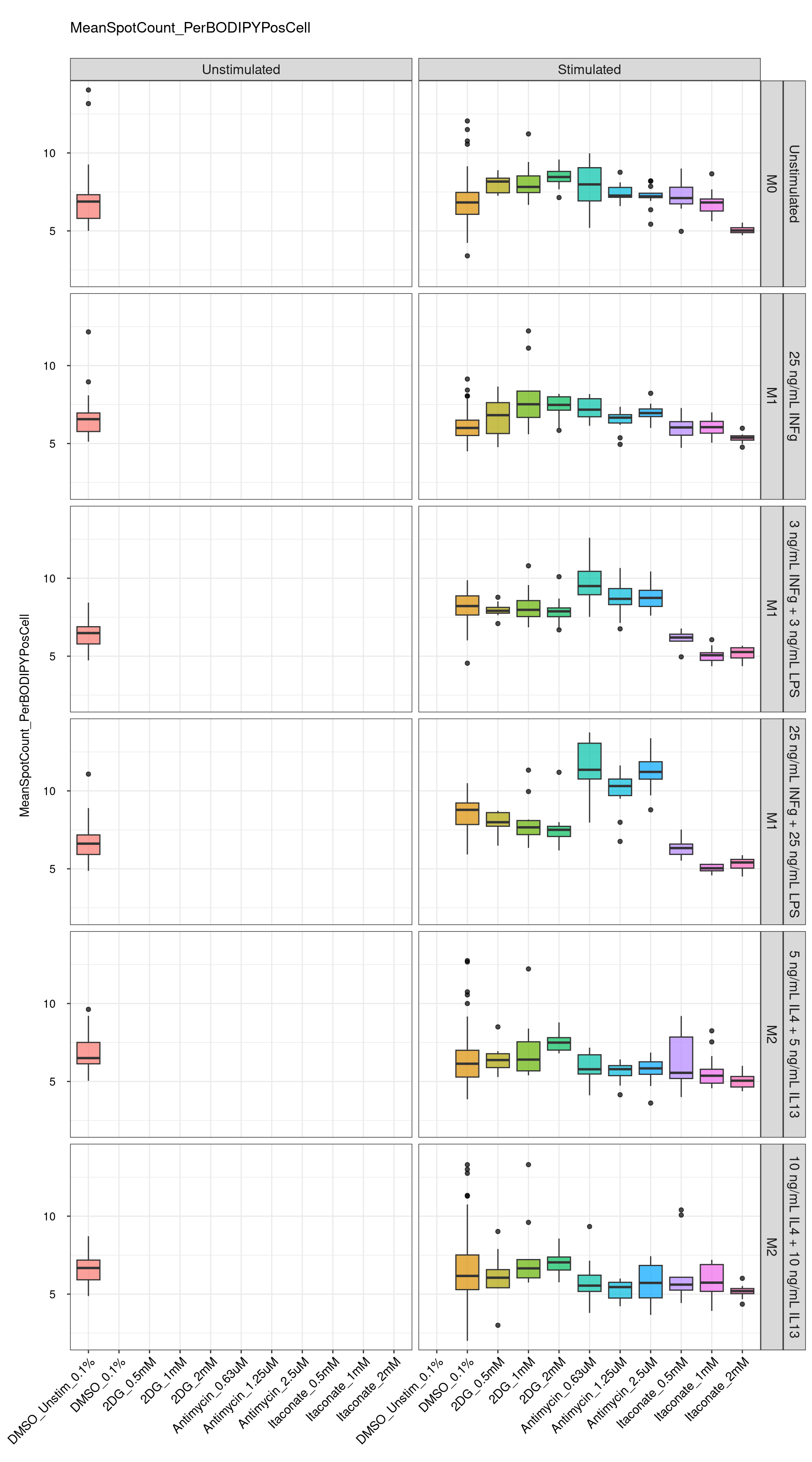
Dot plots
The plots are interactive - toggle particular compounds on and off by clicking the legend, hover over data point on the plot to see its associated value, and hover above the plot on the right-hand side of the screen to see additional options (eg. zoom, select, export).
# Print graphs
for (condition in cond_list){
cat("\n### ", paste0(condition), "{.tabset .tabset-fade .tabset-pills}\n")
cat("\n#### ", value_list[1], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[7], value_list[1], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n##### ", value_list[2], "{.tabset .tabset-fade .tabset-pills}\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[7], value_list[2], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
cat("\n###### ", value_list[3], "\n")
print(htmltools::tagList(plotly::ggplotly(
gen_dotplot(feature_list[7], value_list[3], condition), tooltip = c("Plate", "text"), width = 950, height =350)))
cat("\n")
}Unstimulated
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg
Raw
NormToUnstimDMSO
NormToStimDMSO
3 ng/mL INFg + 3 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
25 ng/mL INFg + 25 ng/mL LPS
Raw
NormToUnstimDMSO
NormToStimDMSO
5 ng/mL IL4 + 5 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
10 ng/mL IL4 + 10 ng/mL IL13
Raw
NormToUnstimDMSO
NormToStimDMSO
Analysed by Ann Rann Wong
Victorian Centre for Functional Genomics
sessionInfo()R version 4.4.2 (2024-10-31)
Platform: x86_64-pc-linux-gnu
Running under: Rocky Linux 9.5 (Blue Onyx)
Matrix products: default
BLAS/LAPACK: FlexiBLAS OPENBLAS-OPENMP; LAPACK version 3.9.0
locale:
[1] LC_CTYPE=en_AU.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_AU.UTF-8 LC_COLLATE=en_AU.UTF-8
[5] LC_MONETARY=en_AU.UTF-8 LC_MESSAGES=en_AU.UTF-8
[7] LC_PAPER=en_AU.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_AU.UTF-8 LC_IDENTIFICATION=C
time zone: Australia/Melbourne
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] magick_2.8.5 lubridate_1.9.4 forcats_1.0.0 stringr_1.5.1
[5] dplyr_1.1.4 purrr_1.0.4 readr_2.1.5 tidyr_1.3.1
[9] tibble_3.2.1 ggplot2_3.5.1 tidyverse_2.0.0 patchwork_1.3.0
[13] viridis_0.6.5 viridisLite_0.4.2 reshape2_1.4.4 DT_0.33
[17] readxl_1.4.4 data.table_1.17.0 workflowr_1.7.1
loaded via a namespace (and not attached):
[1] gtable_0.3.6 xfun_0.51 bslib_0.9.0 htmlwidgets_1.6.4
[5] processx_3.8.6 callr_3.7.6 tzdb_0.4.0 vctrs_0.6.5
[9] tools_4.4.2 crosstalk_1.2.1 ps_1.9.0 generics_0.1.3
[13] parallel_4.4.2 pkgconfig_2.0.3 lifecycle_1.0.4 compiler_4.4.2
[17] farver_2.1.2 git2r_0.35.0 munsell_0.5.1 getPass_0.2-4
[21] httpuv_1.6.15 htmltools_0.5.8.1 sass_0.4.9 lazyeval_0.2.2
[25] yaml_2.3.10 plotly_4.10.4 crayon_1.5.3 later_1.4.1
[29] pillar_1.10.1 jquerylib_0.1.4 whisker_0.4.1 cachem_1.1.0
[33] mime_0.12 tidyselect_1.2.1 digest_0.6.37 stringi_1.8.4
[37] labeling_0.4.3 rprojroot_2.0.4 fastmap_1.2.0 grid_4.4.2
[41] colorspace_2.1-1 cli_3.6.4 magrittr_2.0.3 withr_3.0.2
[45] scales_1.3.0 promises_1.3.2 bit64_4.6.0-1 timechange_0.3.0
[49] rmarkdown_2.29 httr_1.4.7 bit_4.5.0.1 gridExtra_2.3
[53] cellranger_1.1.0 hms_1.1.3 evaluate_1.0.3 knitr_1.49
[57] rlang_1.1.5 Rcpp_1.0.14 glue_1.8.0 vroom_1.6.5
[61] rstudioapi_0.17.1 jsonlite_1.9.1 R6_2.6.1 plyr_1.8.9
[65] fs_1.6.5